
Abstract
Autologous mesenchymal stem cell (MSC) transplants have been used successfully to treat a number of diseases, and patients undergoing cell transplantation must have stem cells collected before transplantation. In this study, we developed a new method to harvest MSCs. Biomaterials were implanted into the spatium intermuscular of mice hind limbs, and a large number of migrating cells (MCs) were isolated from the transplanted biomaterials. The adherent cells in MCs showed the characteristics of MSCs. Further comparative study demonstrated that the characteristics of MC-MSCs were similar to that of bone marrow (BM)-MSCs, including morphology, phenotype, proliferation potential, multilineage differentiation capacity and hematopoiesis-supportive function. The colony-forming unit-fibroblast frequency of the MCs was equivalent to approximately 20-fold of that of the BM. In addition, a BM transplantation experiment demonstrated that MC-MSCs were derived from the peripheral blood. In conclusion, we successfully establish an efficient method to harvest MSCs, and together with the distinct advantages of this method, such as accessibility and possibility for autologous cell therapy, we conclude that our efficient method may be a promising alternative for clinical application.
Keywords
Introduction
Autologous mesenchymal stem cell (MSC) transplants have been used successfully to treat a number of diseases, including musculoskeletal, hematopoietic, neurological and cardiovascular diseases.
1–3
Autologous transplants have the advantage of lower risk of infection during the immune-compromised portion of the treatment, since the recovery of immune function is rapid. Also, the incidence of patients experiencing rejection (graft-versus-host disease) is very rare due to the donor and recipient being the same individual.
1
However, there are limitations to autologous transplants in the collection of stem cells because stem cells must be derived from the patient's own body. Currently, MSCs for autologous transplant are mainly collected from bone marrow (BM).
4,5
Recently, considerable attention in clinical applications has been focused on fat and muscle as an alternative source of MSCs.
3,6,7
However, the methods of harvesting MSCs from these tissues involve tissue biopsy digestion, so they are complicated (using enzymes) and often cause severe tissue damage.
8
Moreover, there are very low frequencies of stem cells in these tissues, and once removed from the body, the proliferation and differentiation potential of cells is limited.
9
Developing an alternative method that could give rise to a better harvest of stem cells is a hot area of research. In this study, we try to establish an efficient method to harvest MSCs for the current clinical applications of gene therapy and tissue engineering. An ideal method of harvesting stem cells for regenerative medicinal applications should meet the following criteria:
Be able to yield high numbers of stem cells; Be a minimally invasive procedure; Stem cells harvested by this method possess a higher proliferation capacity, better cell survival and vigorous capacity for differentiation into multiple tissue lineages; Be easy to operate and in accordance with guidelines for clinical application; Be able to harvest stem cells from patients for autologous transplantation treatment.
Biomaterial provides three-dimensional (3D) support for cell migration, proliferation and differentiation, and thereby acts as a scaffold in a variety of tissue engineering/regenerative medicine applications.
10
Gelatin sponge, because of its flexibility, biocompatibility, biodegradability and porosity, has been applied to the reconstruction of various types of tissues, such as bone, cartilage, heart and blood vessel.
11–13
Muscle, the biggest organ in the human body (comprising approximately 30–40% of total body mass), provides a reservoir for pluripotent stem cells, such as muscle satellite cells,
14
muscle-derived stem cells,
8
multipotent adult progenitor cells,
15
muscle-derived side population cells,
16
endothelial progenitor cells,
17
vasculogenic cells,
17
and so on.
18
Importantly, its extensive network of capillaries facilitates cells from other tissues in the body to migrate into the muscle. Some investigations have shown that pluripotent stem cells in the muscles are derived from the BM.
19
These data suggest that stem cells residing in muscle can be exchanged with those in the BM pool.
20
Moreover, stem cells in the muscles have been identified in the interstitial spaces. We have evidence that the origin of the stem cells may lie within the vascular and perivascular regions in the muscle;
21
recent results also suggest their ultimate origin in blood vessel walls.
22
In addition, it is relatively easy and safe to implant biomaterials into muscle tissue. Therefore, intramuscular implantation is the procedure of choice for collecting stem cells. We previously confirmed that cells captured from spatium intermuscular by porous material exhibited the characteristics of stem cells.
23
Based on this fact, we hypothesized that MSCs in vivo could migrate into biomaterials when biomaterials were implanted into the spatium intermuscular.
To address our hypothesis, in this study, we implanted biomaterials into the spatium intermuscular of mice hind limbs, characterized presumable MSCs in migrating cells (MCs), and carefully analyzed the cell morphology, colony-forming unit-fibroblast (CFU-F) efficiency, phenotype, proliferation and differentiation capacity in vitro, and hematopoiesis-supportive function of the MC-MSCs in comparison with BM-MSCs. In addition, we also elucidated the origin of MSCs in the MCs.
Materials and methods
Animals and ethics
C57BL/c mice, 6–8 weeks of age, were obtained from the Laboratory Animal Center of Shandong University (Jinan, China). All experiments in this study were performed in accordance with the Shandong Academy of Medical Science Animal Care and Use Committee Guide for Laboratory Animals.
Preparation of materials
Gelatin sponges (Gelfoam®) were purchased from Jinling Company (Nanjing, China), and were processed into the wafer with a radius of 2.5 mm, and a thickness of 5 mm, then sterilized by ethylene oxide. All operations were conducted in the laminar flow hood.
Surgical procedure
Mice were put under general anesthesia by light inhalation of diethylether and prepared for surgery. The medial thigh area was disinfected with iodophors. A 1.0-cm midline longitudinal skin incision was made into the medial aspect of the thigh near the body wall. The deep fascia was minimally cut with sterile scissors. One piece of wafer with sterile sponges was prepared by immersion in phosphate-buffered saline (PBS; Gibco, Invitrogen, Grand Island, NY, USA), and then inserted gently into the spatium intermuscular of the exposed muscle group. The skin was closed with interrupted 6-O vicryl sutures.
Sponge weighing and cell counting
At 3, 6, 9, 12, 15 and 18 d after implanting, mice were killed by cervical dislocation. The sponges were retrieved and weighed, and then MCs were collected and counted with a hemocytometer.
Collection of BM and MCs
At 12 d after implanting, the sponges and femurs were taken out and MCs and BM were immediately collected as follows.
BM was collected by flushing the femur with sterile PBS with 2% fetal bovine serum (FBS; Gibco, Invitrogen). The cells were then size-fractionated by being layered onto a Ficoll-Paque (Pharmacia, Piscataway, NJ, USA) gradient and centrifuged for 20 min at 1000
MCs were collected by shredding the sponges with forceps and squeezing the sponge pieces in sterile PBS with 2% FBS. Sponge debris was permitted to settle before the cell-containing supernatant fluid was removed. Cells were washed once and re-suspended in 10 mL of growth medium. Cell numbers were counted with a hemocytometer.
Isolation and culture of adherent cells
The MCs and BM cells were cultured at a density of 1 × 105 cells/cm2 in a 5% CO2 humidified incubator at 37°C. The culture medium was changed to remove non-adherent cells two days after the initial plating. Thereafter, the culture medium was replaced twice each week. The adherent cells were subcultured after the cultures had reached 80–90% confluence; cells were detached by treatment with 0.25% trypsin/ethylenediaminetetraacetic acid (Invitrogen) and were re-plated as passage-1 cells (the process was then continued as previously described).
Colony-forming unit-fibroblast assay
CFU-F assays were performed as described by Yoshimura et al. 24 Briefly, cells from MCs or BM were plated in a 60-cm2 dish in three dilution steps (MCs: 1 × 105/5 × 104/1 × 104 cells/well; BM: 1 × 106/5 × 105/1 × 105 cells/well). Medium was changed every three days. Fourteen days after plating, colonies were subsequently fixed with 4% paraformaldehyde, stained with 0.5% crystal violet in 4% paraformaldehyde for five minutes and washed twice with PBS. The number of colonies was then counted. Colonies of less than 2 mm in diameter and faintly-stained colonies were ignored.
Flow cytometry analysis
To study the phenotypic characteristics, adherent cells (passage 3) from MCs and BM were re-suspended in 2% FBS containing PBS and stained with anti-CD34-fluorescein isothiocyanate (FITC), CD45-FITC, CD29-phycoerythrin (PE; Becton Dickinson, Franklin Lakes, NJ, USA), CD14-FITC, CD44-FITC, CD31-FITC (Serotec, Oxford, UK), SH2, SH3 and SH4 monoclonal antibodies (Osiris Therapeutics, Baltimore, MD, USA) and were analyzed by FACScalibur flow cytometry (Becton Dickinson).
Cell proliferation assay
To study the proliferative ability of the adherent cells derived from MCs and BM, passage 3 cells were seeded at 2 × 105 cells/mL in a 24-well plate and incubated in growth medium. The number of cells was counted in triplicate cultures every day over eight days and growth curves were protracted. The population doubling time (DT) was calculated on the basis of the growth curves using the formula: TD = tlg2/(lgNt − lgN0). N0 is the inoculum cell number, Nt is the cell harvest number and t is the time of the culture (in hours).
Multilineage differentiation assays
To examine the multipotentiality of the adherent cells derived from MC and BM, passage 3 cells were cultured with use of the osteogenesis, chondrogenesis and adipogenesis assay kit (Cyagen, Guangzhou, Guangdong, China) as per the manufacturer's instructions.
Osteogenic differentiation was assessed by incubating the cells with DMEM with 5% FBS supplemented with 10−8 mol/L dexamethasone, 0.2 mmol/L ascorbic acid and 10 mmol/L β-glycerol phosphate for three weeks. To assess calcium deposition, cultures were stained with Alizarin red solution.
Chondrogenic differentiation was assessed by incubation with DMEM with 5% FBS supplemented with 10 ng/mL transforming growth factor-β 3, 100 nmol/L dexamethasone, 50 μg/mL ascorbic acid, 100 μg/mL sodium pyruvate, 40 mg/mL proline and Insulin-Transferrin-Selenium-plus (Gibco, Invitrogen) at the final concentrations (6.25 μg/mL bovine insulin, 6.25 μg/mL transferrin, 6.25 μg/mL selenious acid, 5.33 μg/mL linoleic acid and 1.25 μg/mL bovine serum albumin) for three weeks. To assess formation of sulfated proteoglycan-rich matrix, cells were stained with fresh Alcian blue solution.
Adipogenic differentiation was assessed by incubation with DMEM with 5% FBS supplemented with 0.5 μmol/L hydrocortisone, 0.5 μmol/L isobutylmethylxanthine and 60 μmol/L indomethacin for three weeks. To assess accumulation of neutral lipid vacuoles, cells were stained with fresh Oil red O solution.
Long-term hematopoietic supportive assay
Sponges were implanted into male mice. At 12 d after implanting, sponges and femurs were taken out, and MC-MSCs and BM-MSCs were immediately isolated by the method described above and counted. Mononuclear cells (MNCs) were isolated from the BM of female mice by the method described previously and counted. 25,26 Forty-five female C57BL/c mice weighing 18–22 g were irradiated with 8.5 Gy, and then randomly divided into three groups with 15 mice each. Group 1 was injected on day 2 after irradiation with 0.2 mL of 1.0 × 107 MNCs. Group 2 was injected on day 2 after irradiation with 0.2 mL of 1.0 × 107 MNCs plus MC-MSCs, 0.5 × 107, respectively. Group 3 was injected on day 2 after irradiation with 0.2 mL of 1.0 × 107 MNCs plus BM-MSCs, 0.5 × 107, respectively. Eight weeks later, BM samples were collected from the recipient and used for Y chromosome polymerase chain reaction (PCR), and then BM samples were cultured on methylcellulose-based medium (Methocult; Stem Cell Technologies, Vancouver, BC, Canada) at 37°C for two weeks; hematopoietic colonies greater than 50 cells were counted.
BM transplantation
To determine the source of MSCs in the MCs, we performed BM transplantation. Male-derived BM were obtained and transplanted intravenously into female mice, and then sponges were implanted into the spatium intermuscular of recipient's hind limbs. Sponges were taken out on day 12 after implantation, MSCs were isolated and cultured as described above, and then evaluated by Y chromosome PCR (see below).
Y chromosome PCR
The Sry gene of the Y chromosome in the BM and MC-MSC of recipient were detected by PCR. The mouse Y-chromosome-specific Sry primers were: (sense primer) 5′ ATTTATGGTG TGGTCCCG 3′; (anti-sense primer) 5′ GCTGTAAAATGCCACTCC 3′. The PCR conditions were: 36 cycles at 95°C for 50 s, 55°C for 60 s, 72°C for 60 s, followed by one cycle at 72°C for 10 min. All PCR amplifications included a male (positive) and female (negative) control. All experiments were run in triplicate.
Statistics
The results were expressed as mean ± standard deviation (SD). Significant differences between means were determined by Student's t-test. A P value less than 0.05 was considered statistically significant.
Results
Sponge weight and cell number
The sponges were weighed, and the MC number counted at 3, 6, 9, 12, 15 and 18 d after implantation (Figure 1). On the basis of the curves, sponge weights were slightly reduced within 12 d after implanting, but deeply decreased after 12 d, showing a serious sponge degradation after 12 d (Figure 1a). However, MC numbers significantly increased, and reached the maximum after 12 d (Figure 1b). We therefore suggest that the 12th day after implanting is a suitable time to retrieve the sponges.
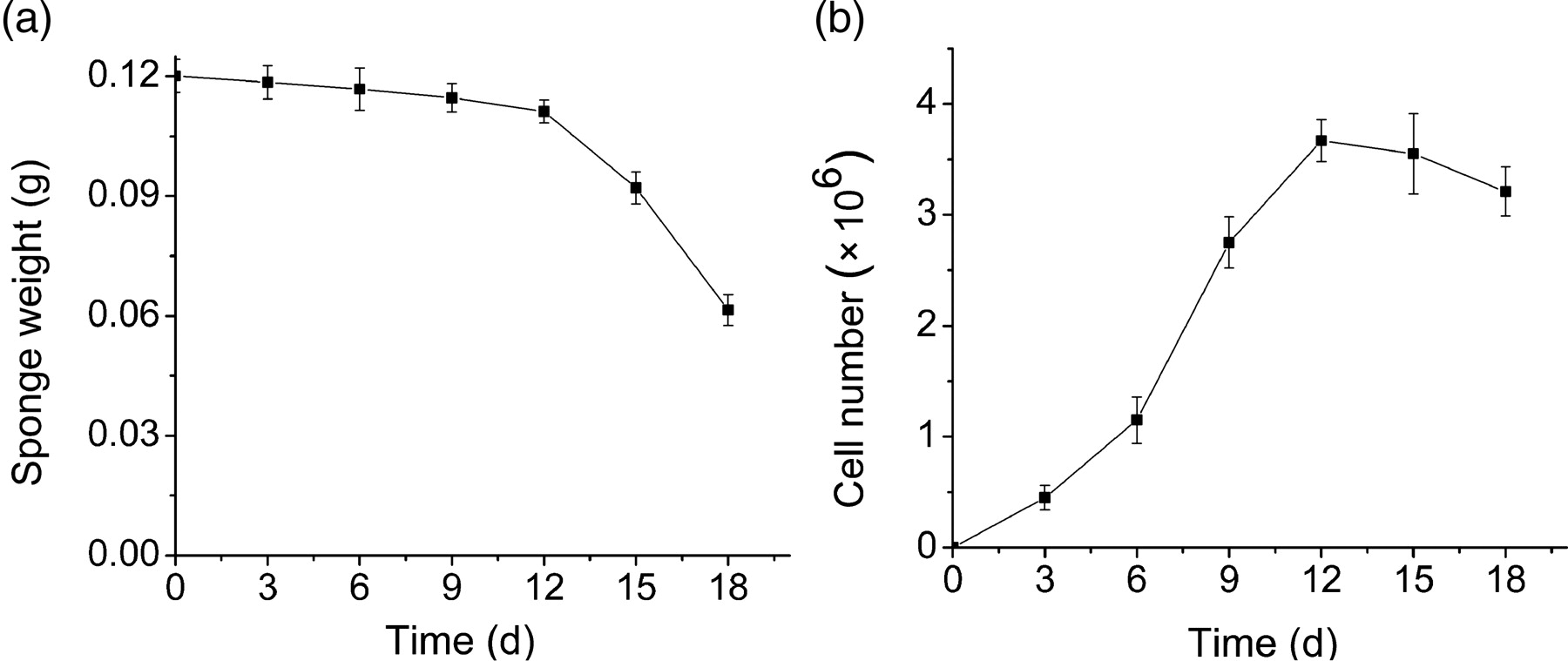
The sponges were weighed and migrating cell (MC) number was counted on 3, 6, 9, 12, 15 and 18 d of implantation. (a) The weight-for-time curve of sponges. Points represent mean ± SD. (b) The number-for-time curve of MCs. Points represent mean ± SD
Morphology characteristics
On day 7 after initial plating, both MC- and BM-derived cells adhered to the plastic surface and presented a small population of single cells with a spindle shape. The adherent cells looked like long, spindle-shaped, fibroblastic cells, began to form colonies and confluent. No obvious differences in morphology were noted between the two populations (Figure 2a).
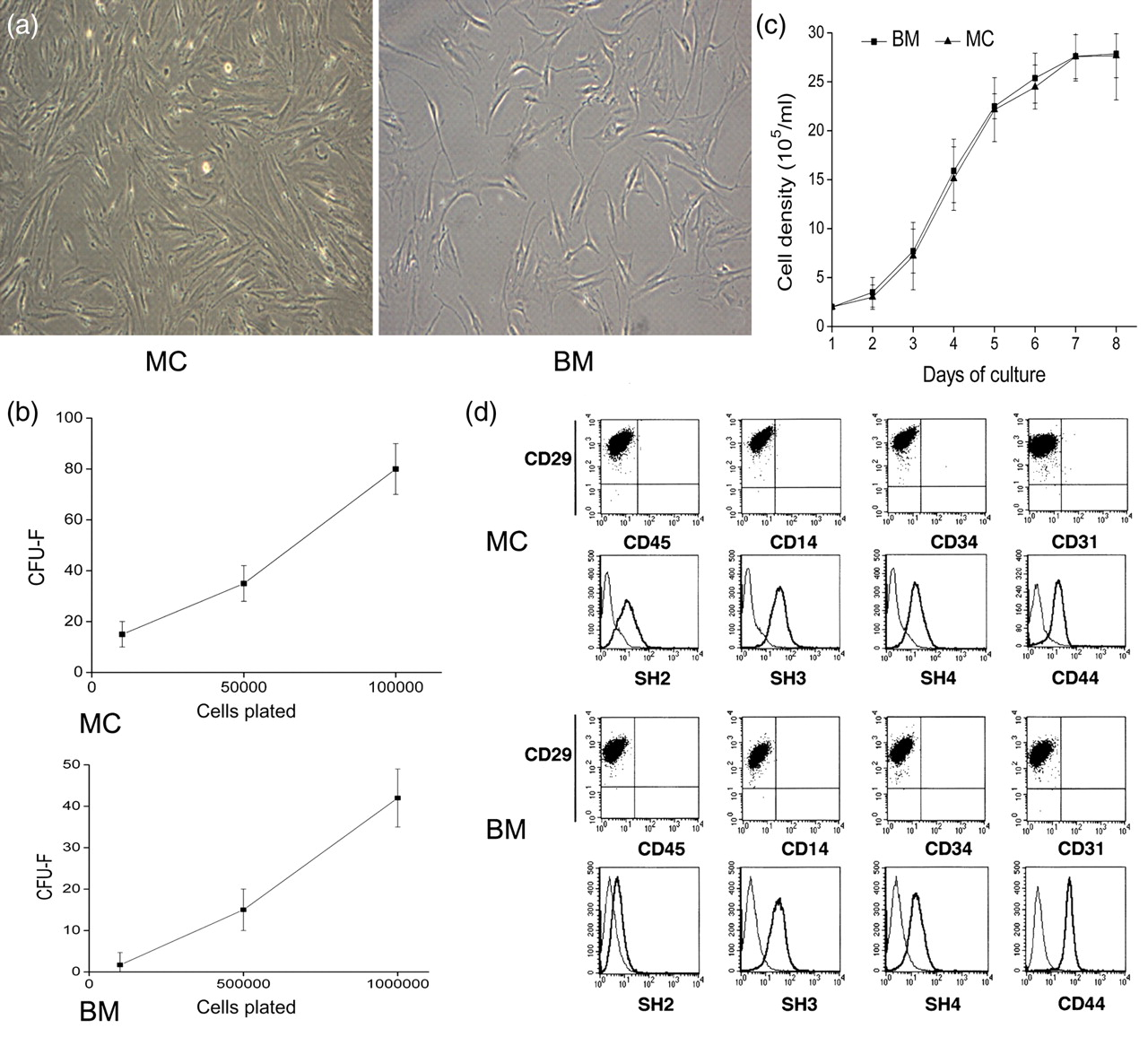
Morphology, colony-forming unit-fibroblast (CFU-F), phenotype, and proliferation potential of migrating cell (MC)- and bone marrow (BM)-derived adherent cells. (a) Morphology of MC- and BM-derived adherent cells. After the initiating plate on day 7, adherent cells derived from MCs displayed a fibroblastic morphology similar to that of the adherent cells from BM (magnification ×100). (b) Frequency of CFU-F in MCs and BM. (c) Growth curves of MC- and BM-derived adherent cells. Similar growth patterns were observed. The average population doubling time was calculated on the basis of the growth curves. (d) Immunophenotype of adherent cells by fluorescence-activated cell sorting (FACS) analysis. Adherent cells derived from MCs and BM were stained with surface antibodies and analyzed by a FACS sorter. (A color version of this figure is available in the online journal)
CFU-F efficiency
To compare the contents of stem cells in MCs and BM, CFU-F was analyzed. Figure 2b shows numbers of CFU-F colonies at different cell-seeding densities for both MC and BM-derived cells, which confirmed a higher frequency of CFU-F in MCs (82.2 ± 10.6/105) than in BM (43.7 ± 7.4/106). Also, Figure 2b shows that the frequency of CFU-F increased cell-seeding densities for both MC- and BM-derived cells.
Proliferative potential
Growth curves depicted an initial lag phase of one day, followed by a log phase in which cells divided at exponential rates for 2–6 d. The log phase was followed by a plateau phase. Adherent cells derived from MCs could be readily expanded in vitro by successive cycles of trypsinization, seeding and culture every three days for 20 passages. Cells that had undergone up to 20 passages displayed no visible changes either in terms of their morphology by light microscopy. Similar growth patterns were observed with adherent cells derived from MCs and BM (Figure 2c). The mean cumulative time of population doublings of MC and BM adherent cells was 22.6 ± 3.5 and 21.3 ± 5.3 h, respectively. These data indicated that MC- and BM-derived adherent cells both had a similar proliferation capacity.
Phenotypic characterization
We examined the surface marker profile of the adherent cells derived from MCs and BM using FACS. The phenotype of the adherent cells derived from MCs was similar to that of adherent cells derived from BM. These cells were positive for CD29, CD44, SH2, SH3 and SH4 but were negative for CD14, CD31, CD34 and CD45 (Figure 2d).
Multilineage differentiation potential
Osteogenic, chondrogenic and adipogenic differentiation of adherent cells derived from MCs and BM were examined by specific cytohistological staining. The representative pictures are shown in Figure 3. After three weeks of induction in conditioned medium, both adherent cells derived from MCs and BM were capable of differentiation into osteocytes, chondrocytes or adipocytes, shown by positive staining of Alizarin red (Figures 3a and d), Alcian blue (Figures 3b and e) or Oil red O (Figures 3c and f). No apparent differences in the osteogenic, chondrogenic and adipogenic differentiation capacity were detected between BM- and MC-derived adherent cells.
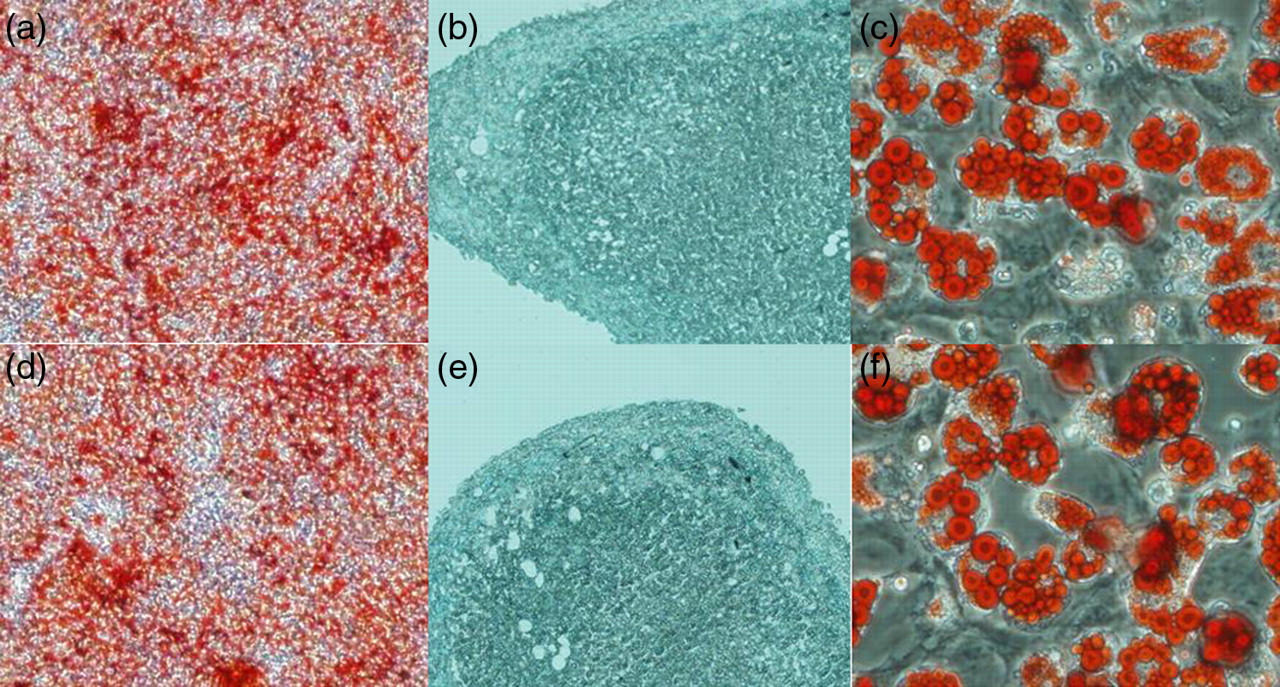
Multilineage differentiation of adherent cells derived from migrating cells (MCs) and bone marrow (BM) in vitro. MCs (a–c) and BM (d–f) into osteocytes, chondrocytes and adipocytes. Osteogenic differentiation was indicated by calcium deposition (a and d, ×40 magnification), which stained with Alizarin red. Chondrogenic differentiation was shown by the formation of sulfated proteoglycan-rich matrix, which stained with Alcian blue (b and e, ×200 magnification). Adipogenic differentiation was indicated by accumulation of neutral lipid vacuoles that stained with Oil red O (c and f, ×200 magnification). (A color version of this figure is available in the online journal)
Detection of MSC engraftment
The BM of the recipients from groups 2 and 3 were demonstrated harboring a DNA fragment, 239 bp in length, of the Y chromosome Sry region of donor mice, indicating that MC-MSCs and BM-MSCs could migrate into BM and survive in the irradiated mice (Figure 4a).
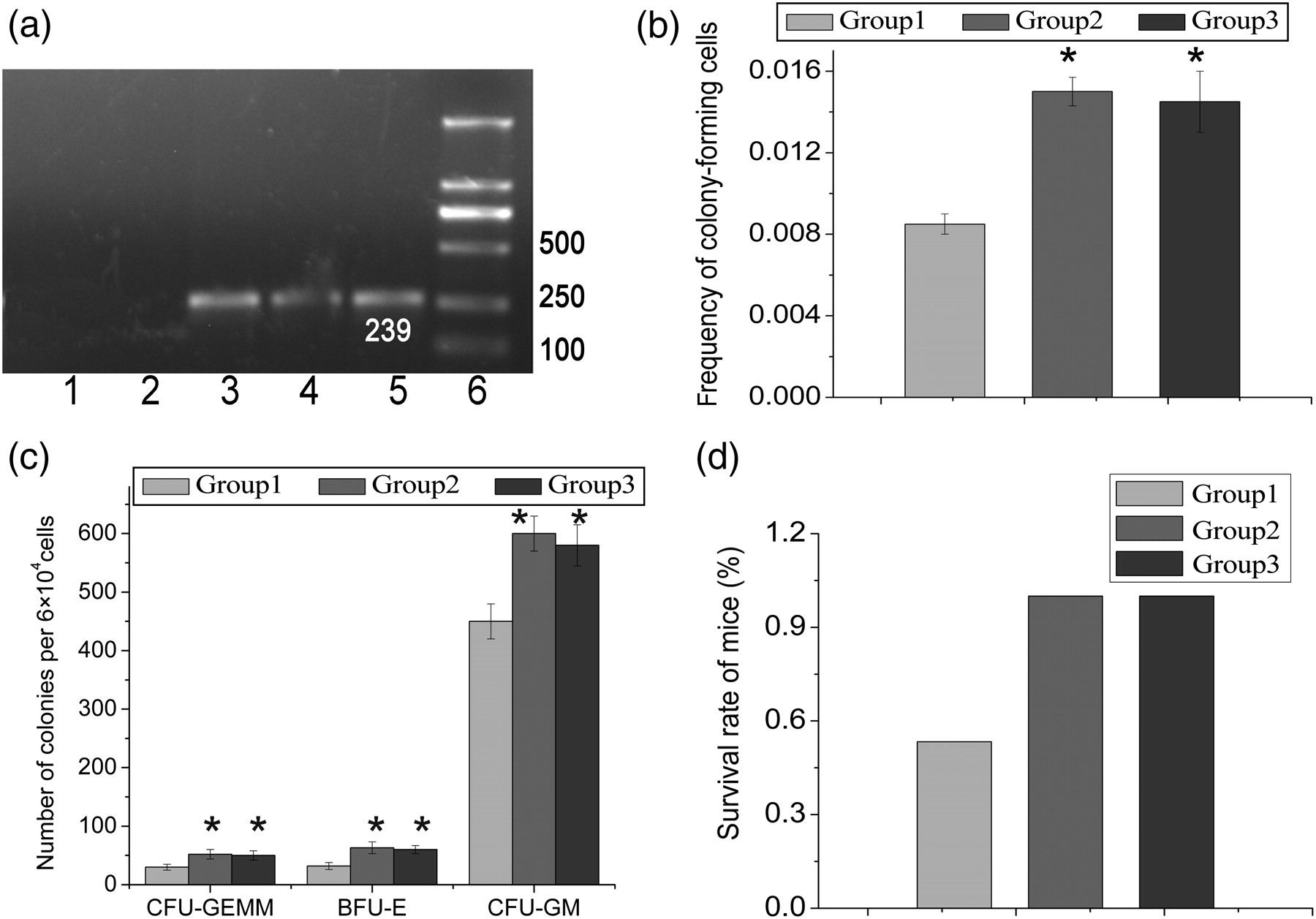
Hematopoiesis-supportive function assay. (a) Mesenchymal stem cells (MSCs) in the migrating cells (MCs) could home to the bone marrow (BM) of irradiated recipients. DNA samples were extracted from the BM. Male-derived cells were evaluated by polymerase chain reaction. Lane 1: female, lane 2: group 1, lane 3: group 2, lane 4: group 3, lane 5: male, lane 6: DNA marker. (b) Comparisons of the percentages of colony-forming unit (CFU) from the BM of recipients after co-transplantation of mononuclear cells (MNCs) (control), MNCs/MC-MSCs or MNCs/BM-MSCs. Bars indicate mean ± SD (n = 5 per each group). *P < 0.05 versus control. (c) Comparisons of the numbers of each type of colony in each group. Bars indicate mean ± SD (n = 5 per each group). *P < 0.05 versus control. (d) The survival rates of mice from each group
Hematopoietic assay
In co-transplantation of MNCs/MC-MSCs or MNCs/BM-MSCs, the frequency of CFU was significantly higher (P < 0.05) than those in MNC transplantation alone (Figure 4b). Moreover, the proportion (BFU-E:CFU-GM:CFU-GEMM; BFU-E, burst-forming unit-erythroid; CFU-GM, colony-forming unit-granulocyte/macrophage; CFU-GEMM, colony-forming unit-granulocyte/erythroid/macrophage/megakaryocyte) of each colony type was proportionally increased compared with transplantation with MNCs alone (Figure 4c). In addition, there was no significant difference in numbers of BFU-E, CFU-GM or CFU-GEMM between MNC/MC-MSC and MNC/BM-MSC co-transplantation (P > 0.05). These findings indicate that MC- and BM-derived MSCs both have a similar hematopoiesis-supportive function.
Rescue of irradiated mice
In the set of experiments in which cells were injected into irradiated mice, 8/15 mice from group 1 survived for over two months. In contrast, all mice from groups 2 and 3 have survived for over two months (Figure 4d). This result shows that MC- and BM-derived MSCs both have a similar hematopoiesis-supportive function.
Source of MSCs in the MCs
To examine whether the MSCs in the MCs were derived from the circulating peripheral blood (PB), cell samples on day 12 post-transplantation were evaluated by Y chromosome PCR (Figure 5a). The samples were demonstrated harboring a DNA fragment, 239 bp in length, of the Y chromosome Sry region of donor mice (Figure 5b), indicating that MSCs in the MCs originated from the PB.

Mesenchymal stem cells (MSCs) in the migrating cells (MCs) originated in the peripheral blood (PB). (a) Experimental scheme. The female mice underwent transplantation with 5 × 106 male-derived bone marrow (BM) cells. At 24 h after transplantation, sponges were implanted into the spatium intermuscular of mice hind limbs. The MCs were sampled and analyzed at 12 d after implantation. (b) MSCs in the MCs from recipients were evaluated by Y chromosome polymerase chain reaction. Lane 1: female, lane 2: MC-MSCs, lane 3: male, lane 4: DNA marker
Discussion
In the present study, we successfully implanted sponges into the spatium intermuscular of mice hind limbs and collected a large number of cells in the sponges after 12 d. We speculated that this might be due to absorption of gelatin sponge and cell migration and proliferation effects. Gelatin sponge absorption and cell migration played an important role in the early period after implantation. When adsorption capacity of the sponge was lost, stem cells that previously colonized the sponge would proliferate in response to stimulation of hypoxia, collagen and inflammatory factors. The surgical procedure of implanting biomaterials did not cause traumatic bleeding, but induced a classic inflammatory response, so we speculated that some inflammatory cells penetrated into biomaterials in the early days. When the sponges were retrieved at 12 d after implantation, no neo-vasculature or vasculature-like structures formed around or within the gelatin sponge. We isolated MCs and found some inflammatory cells in MCs; we thought that these inflammatory cytokines played an important role in MSC migration and proliferation. Some cells might enter a decline phase after the log phase and plateau phase. In addition, degradation of the gelatin sponge might also be a reason for the decrease in MC numbers after 12 d of implantation.
Previously, we also implanted polylactic acid (PLA) into the spatium intermuscular, and similar numbers and proportions of cells were harvested as when sponges were implanted. However, compared with PLA, the sponge exhibits better flexibility, biocompatibility and affinity to cells, 12 so we concluded that sponge would be an excellent candidate for this study. We isolated cells from sponges by a manual method, as most cells can be isolated by this method. Previously, we also isolated MCs using a digestion method. Sponges were digested with collagenase to release cells, and similar numbers and proportions of cells were collected as they were when sponges were merely minced and rinsed. This indicated that all or almost all MCs were harvested by a manual method. Compared with the digestion method, the manual method is more suitable for the next study, as this method is not only simple to operate (diminished time) but also has less effects on cell activity.
Stem cells are usually isolated and purified from a variety of sources by way of their physical propensity to adhere to the plastic substrate of the cell culture plate.
26,27
In this study, we isolated and purified stem cells from MCs using this method and explored their biological characteristics. The International Society for Cellular Therapy has also provided the following minimum criteria for defining multipotent mesenchymal stromal cells:
4
Plastic-adherent under standard culture conditions; Positive for expression of CD105, CD73, CD29, CD44 and CD90, and absent for expression of hematopoietic cell surface markers CD34, CD45, CD14, and CD31; Under specific stimulus, cells should differentiate into osteocytes, adipocytes and chondrocytes in vitro.
Our results suggest that MC-derived stem cells shared the general biological characteristics of MSCs. Further comparative study demonstrated that the characteristics of MC-MSCs were similar to that of BM-MSCs, including morphology, phenotype, proliferation potential and multilineage differentiation capacity,
4
but the estimated amount of MSCs in total MCs would be equivalent to approximately 20-fold of that of MSCs in the total BM. In addition, MC-MSCs did not express myoblast phenotype MyoD (data not shown). MyoD, as one of the earliest markers of myogenic commitment, is expressed at an earlier stage of myogenesis when quiescent stem cells rapidly expand in number to generate the myoblasts needed to repair tissue damage, but not in quiescent stem cells.
28
This suggested that the surgical procedure of implanting biomaterials did not cause muscle damage.
MSCs have been implicated as playing an important role in hematopoietic stem cell engraftment. MSCs are capable of improving HSC engraftment and accelerating hematopoietic reconstitution based on the hematopoiesis-supporting ability. 29,30 We demonstrated that MC- and BM-derived MSCs both had similar hematopoiesis-supportive function, indicating that MCs could be considered a promising alternative in the field of HSC transplantation to BM as a source of MSCs.
In addition, we demonstrated that MSCs in the MCs originated from the PB. The MSCs in the BM constantly move from the BM to systemic circulation and home to the BM and extramedullary tissues, including muscle, spleen and adipose tissue, and they are also able to migrate and home to sites of inflammation following tissue injury, 31,32 by an identified mechanism. In this study, the surgical procedure of implanting biomaterials did not cause traumatic bleeding. There are four possibilities for MSCs migrating into biomaterials: (1) biomaterial constitutes an hypoxic environment, which can provide suitable oxygen tension for MSCs; (2) muscle is supported by unique, but well-organized, vascular systems. 23 The hypoxic environment and characteristic vascular system may play important roles in the recruitment and regulation of MSCs; 4,33,34 (3) biomaterial has a 3D porous structure that can largely facilitate in vivo cell migration and penetration; and (4) implanted biomaterial induces a classic inflammatory response, with MSCs to infiltrate the biomaterial. Based on these possibilities, we propose that MSCs in the PB could mobilize to biomaterial and maintain their properties and function properly.
In summary, we provide an efficient method for harvesting MSCs by implanting biomaterials into the spatium intermuscular of mice hind limbs. To the authors' knowledge, this is the first report of this technique. Unlike the current methods, our new method is not only relatively less invasive and causes less tissue and functional damage, but it is also simple to operate (does not use enzyme). In addition, it is possible for autologous cell therapy. We believe that implantation of biomaterial could be practically used as an alternative method to harvest MSCs for clinical applications.
Footnotes
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (grant nos. 30940065 and 30571766) and the Shandong Province Natural Science Foundation (grant no. Y2008C06).