
Abstract
Pregnancy is characterized by physiological adjustments in the maternal compartment. In this investigation, the influence of pregnancy on maternal liver was examined in CD-1 mice. Dramatic changes were observed in the size of the maternal liver during pregnancy. Livers doubled in weight from the non-pregnant state to day 18 of pregnancy. The pregnancy-induced hepatomegaly was a physiological event of liver growth confirmed by DNA content increase and detection of hepatocyte hyperplasia and hypertrophy. Growth of the liver was initiated following implantation and peaked at parturition. The expression and/or activities of key genes known to regulate liver regeneration, a phenomenon of liver growth compensatory to liver mass loss, were investigated. The results showed that pregnancy-dependent liver growth was associated with interleukin (IL)-6, tumor necrosis factor α, c-Jun and IL-1β, but independent of hepatocyte growth factor, fibroblast growth factor 1, tumor necrosis factor receptor 1, constitutive androstane receptor and pregnane X receptor. Furthermore, maternal liver growth was associated with the activation of hepatic signal transducer and activator of transcription 3, β-catenin and epidermal growth factor receptor, but pregnancy did not activate hepatic c-Met. The findings suggest that the molecular mechanisms regulating pregnancy-induced liver growth and injury-induced liver regeneration exhibit overlapping features but are not identical. In summary, the liver of the mouse adapts to the demands of pregnancy via a dramatic growth response driven by hepatocyte proliferation and size increase.
Introduction
Pregnancy induces widespread maternal changes in the structures and functions of virtually every organ system, including the brain, 1 pancreas, 2–4 immune system 5 and cardiovascular system. 6 These physiological changes represent essential maternal adaptations to meet the needs of the development and growth of the placenta and fetus. Understanding the physiology of maternal adaptive responses to pregnancy is of particular importance to gain insights into pregnancy-associated diseases. 7
Maternal liver exhibits profound changes during pregnancy. Maternal hepatic functional adjustments include alterations in drug, lipid, cholesterol, bile acid and glucose metabolism. 8–24 Pregnancy-induced changes in liver function tests have been reported in humans, including alterations of serum albumin and bilirubin levels, aspartate transaminase and alanine transaminase activities, and triglyceride and cholesterol concentrations in the blood. 25–29 Remarkably, the maternal liver responds to pregnancy by significantly increasing its size. 8,11,30–33 Although this phenomenon was first reported over 50 years ago, few efforts have attempted to elucidate mechanisms regulating pregnancy-induced liver growth. Our group revealed that maternal liver enlargement during pregnancy is a growth response induced by hepatocyte proliferation and also identified concomitant changes in hepatic gene expression. 34 Williamson and co-workers 35 provided insights into potential roles of reproductive hormones, bile acids and the nuclear receptors mediating bile acid signaling in the regulation of pregnancy-induced hepatomegaly.
In this investigation, we examined maternal adjustments to pregnancy from embryo implantation to parturition in the mouse. Maternal hepatic growth responses to pregnancy are characterized by both hepatocyte hyperplasia and hypertrophy. Some mechanisms controlling maternal liver growth are shared with mechanisms controlling injury-induced liver regeneration, while others are distinct.
Materials and methods
Animals and tissue preparations
CD-1 mice were purchased from Charles River Laboratories, Inc (Wilmington, MA, USA). Tumor necrosis factor receptor 1 (TNFR1)-null mice on a C57BL/6 background were provided by Drs Kathy F Roby and Paul F Terranova (University of Kansas Medical Center, Kansas City, KS, USA). 36 C57BL/6 mice were obtained from Harlan Inc (Indianapolis, IN, USA). The animals were housed in an environmentally controlled facility, with lights on from 06:00 to 20:00 h, and allowed free access to food and water. Timed pregnancies were generated and the presence of a copulatory plug in the vagina was designated as gestation day 1. Non-pregnant, pregnant and postpartum non-lactating mice were weighed and sacrificed. Livers and kidneys were dissected and weighed. Liver tissues were fixed in formalin and then embedded in paraffin for histological analysis, or snap-frozen in liquid nitrogen for total RNA isolation and biochemical analysis. Litter sizes for pregnancies ranged from 8 to 14. Protocols for the care and use of animals were approved by the University of Kansas Medical Center Animal Care and Use Committee.
Measurements of total hepatic DNA content
Liver tissues were completely digested overnight at 55°C in a buffer containing 100 mmol/L NaCI, 50 mmol/L Tris-HCI (pH 8.0), 25 mmol/L EDTA (pH 8.0), 0.5% SDS and proteinase K (400 μg/mL). The digested samples were mixed with PicoGreen dsDNA Quantitation Reagent (Molecular Probes, Eugene, OR, USA). The mixed samples were excited at 480 nm and the fluorescence emission intensity was measured at 520 nm using a spectrofluorometer. DNA concentrations were determined with a standard curve of fluorescence emission intensity versus DNA concentration.
Ki-67 immunostaining and mitotic figure and hepatocyte counting
Formalin-fixed and paraffin-embedded liver samples were sectioned and subjected to Ki-67 immunostaining with a primary Ki-67 antibody (NeoMarkers, Fremont, CA, USA) according to the manufacturer's instructions. Ki67-positive hepatocytes and mitotic figures were counted in five low magnification microscopic fields of ×40 and ×20, respectively, for each sample. Formalin-fixed and paraffin-embedded liver sections were stained with hematoxylin and eosin (H&E). Hepatocytes were counted with Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA) in at least four low-magnification (×40) microscopic fields for each sample.
Quantitative realtime polymerase chain reaction
Total RNA was isolated from frozen liver tissue using the TRIzol reagent according to the manufacturer's protocol (Invitrogen, Carlsbard, CA, USA). The cDNAs were synthesized with total RNA (1 μg) from each sample, diluted 10 times with water and subjected to quantitative realtime reverse transcriptase polymerase chain reaction (qRT-PCR) to quantify mRNA levels using TaqMan probe (Applied Biosystems, Foster City, CA, USA). Primers and probes were designed using Primer Express 2.0 (Applied Biosystems). The probe was labeled with a reporter dye FAM (6-carborxyfluorescein). TaqMan Universal PCR Master Mix (Applied Biosystems) was used to prepare the PCR incubations. Primers and probes were added at a final concentration of 90 and 125 nmol, respectively, in a total volume of 20 μL. The amplification reactions were carried out in the ABI Prism 7900 sequence detection system (Applied Biosystems) with initial hold steps (50°C for 2 min, followed by 95°C for 10 min) and 40 cycles of a two-step PCR (92°C for 15 s, 60°C for 1 min). The primer (Integrated DNA Technologies, Coralvillem, IA, USA) and probe (Sigma, St Louis, MO, USA) sequences used for qRT-PCR are listed in Table 1. The comparative cyclic threshold (CT) method was used for relative quantification of the amount of mRNA for each sample normalized to β-actin transcript levels.
Sequences of primers and probes for quantitative realtime polymerase chain reaction
IL, interleukin; TNF, tumor necrosis factor; HGF, hepatocyte growth factor; PXR, pregnane X receptor; CAR, constitutive androstane receptor
Northern blot analysis
Northern blot analysis was performed as previously described. 37 Briefly, total RNA was extracted from frozen liver tissues using Trizol reagent (Invitrogen). Total RNA (20 μg) was separated on 1% formaldehyde agarose gels and transferred onto nylon membranes. Blots were probed with a [32P]-labeled cDNA for fibroblast growth factor 1 (FGF1). A glyceraldehyde-3-phosphate dehydrogenase (G3PDH) cDNA was used as an internal reference to ensure equal loading and RNA integrity.
Western blot analysis
Liver homogenates (40 μg) were separated by polyacrylamide gel electrophoresis under reducing conditions. Proteins from the gels were electrophoretically transferred on to nitrocellulose. Antibodies of signal transducer and activator of transcription 3 (STAT3), epidermal growth factor receptor (EGFR), p-EGFR, cyclin E and cyclin A2 from Epitomics (Burlingame, CA, USA); p-STAT3 (Tyr 705), β-catenin, cyclin D1, caspase 3 and non-phosphorylated β-catenin from Cell Signaling Technology (Danvers, MA, USA); c-Met and p-c-Met from Abcam (Cambridge, MA, USA); and β-actin from Santa Cruz Biotechnology (Santa Cruz, CA, USA) were used as probes. Immune complexes were detected using the enhanced chemiluminescence system (Pierce, Rockford, IL, USA).
Statistical analysis
Data are shown as mean ± standard error of the mean (SEM) or standard deviation (SD). Statistical analyses were performed using one-way analysis of variance. Comparisons of means were determined by post hoc analyses. Significant differences were defined when P < 0.05.
Results
Pregnancy induces maternal liver mass expansion
Gravimetric responses of maternal livers and kidneys were assessed throughout pregnancy and during postpartum (day 10, non-lactating). As expected, pregnancy-dependent increases in maternal liver wet weight (hepatomegaly) were observed (Figure 1a). In comparison with prepregnancy liver sizes, the maternal liver mass expansion became statistically significant at gestation day 13 and was sustained until the end of pregnancy. When compared with liver weights at gestation day 11 (1.73 ± 0.23 g), liver weights at gestation days 13 (2.26 ± 0.28 g), 15 (2.48 ± 0.39 g) and 18 (2.62 ± 0.49 g) were significantly higher (P < 0.05). Thus, robust maternal liver mass expansion occurred during the second half of pregnancy. After parturition, the maternal liver regressed to prepregnancy sizes. Two waves of significant elevation of the liver-to-body weight ratios were seen during the course of gestation (Figure 1b). The first wave occurred during the first half of pregnancy (gestation days 8–10), whereas the second wave took place during the second half of gestation (gestation days 13–15). The postpartum liver exhibited a higher liver-to-body weight ratio than determined for non-pregnant mice. In contrast to the liver, there were no significant maternal kidney weight changes during pregnancy (Figure 1c). Significant decreases in kidney-to-body weight ratio during the second half of pregnancy were caused by body weight gain (Figure 1d). The results demonstrate that pregnancy induces maternal hepatomegaly, which is not generalized to other maternal organs.
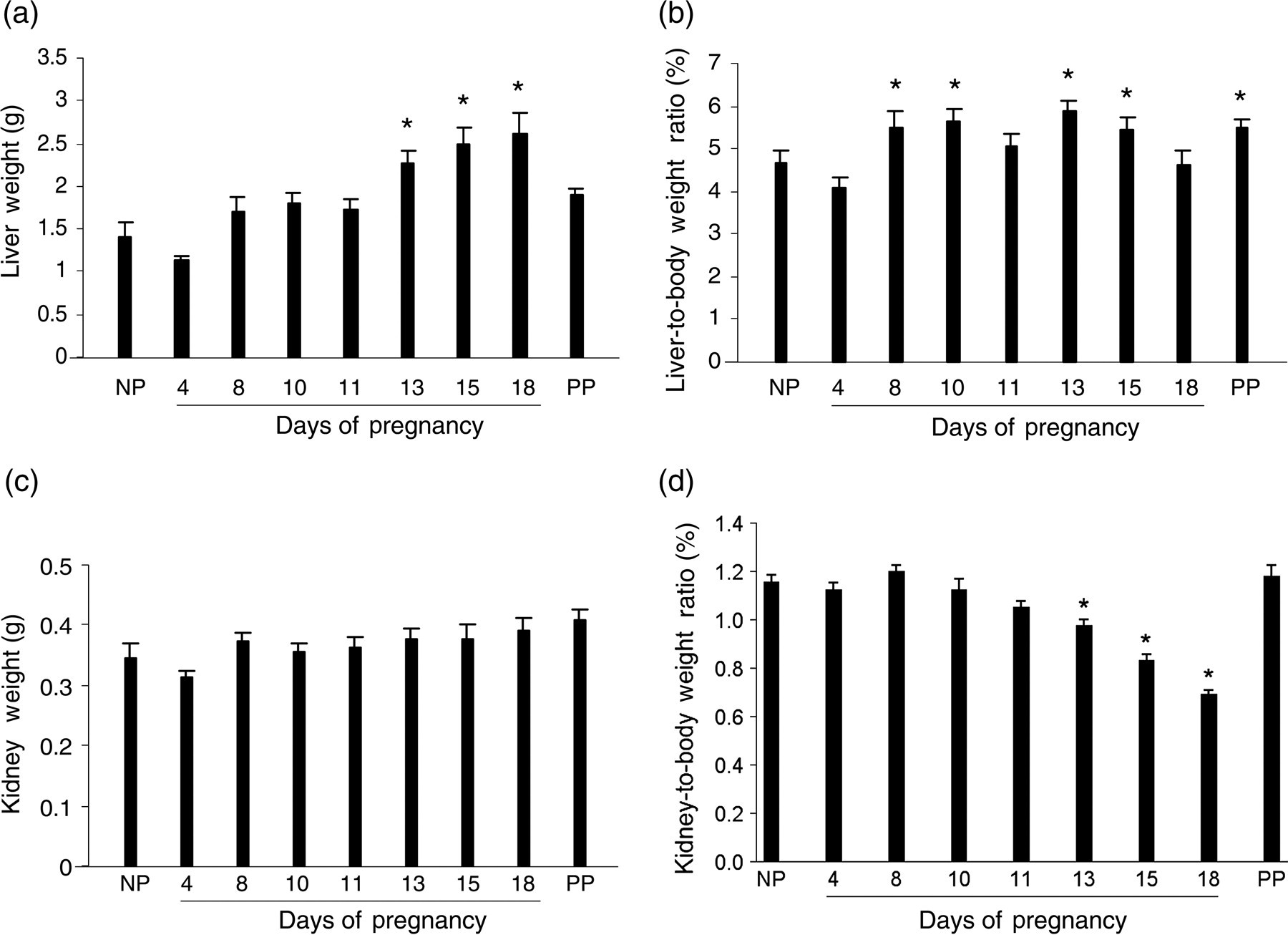
Maternal liver and kidney weight responses to pregnancy. Livers and kidneys were collected and weighed from non-pregnant (NP), pregnant (gestation days 4, 8, 10, 11, 13, 15 and 18) and day 10 postpartum (non-lactating; PP) CD-1 mice. Liver weights (a), liver-to-body weight ratios (b), kidney weights (c) and kidney-to-body weight ratios (d) are presented. Data are expressed as mean ± SEM (n = 5). Asterisks indicate P < 0.05 in comparison with non-pregnant mice. Note that the liver, but not kidney, responds to pregnancy by net weight gain and increase of liver-to-body weight ratio
Pregnancy-induced maternal liver mass increase is driven by hepatocyte hyperplasia and hypertrophy
To determine whether liver size increase is the result of hyperplasia and/or hypertrophy of hepatocytes, we first measured the total DNA content in the livers of non-pregnant, pregnant (gestation days 8, 13 and 18) and day 10 postpartum (non-lactating) CD-1 mice. Liver DNA content was significantly increased on gestation days 13 and 18 as compared with non-pregnant mice (Figure 2). Postpartum and non-pregnant mouse liver DNA contents did not significantly differ. The changes of hepatic total DNA content resembled the gestational pattern of liver weight increase (Figure 1), suggesting that liver cell proliferation may occur during pregnancy. To confirm this, we performed Ki-67 immunostaining using the livers of non-pregnant and pregnant mice to visualize cell proliferation. The numbers of Ki67-positive hepatocytes were markedly increased on gestation days 8, 10, 13, 15 and 18 in comparison with that in non-pregnant mice (Figures 3a and b). The increases in the numbers of mitotic figures of hepatocytes, an indicator of hepatocytes undergoing cell division, resembled the changes in the numbers of Ki67-positive hepatocytes during the course of pregnancy (Figures 3b and c). These findings demonstrate that hepatocyte proliferation contributes to maternal liver mass expansion. To estimate whether liver cell hypertrophy is also a mechanism underlying maternal liver enlargement, hepatocytes were counted per microscopic field in livers from non-pregnant and pregnant mice. The results showed that the numbers of hepatocytes per microscopic field significantly decreased especially during the second half of gestation, compared with observations from non-pregnant mice (Figure 4). The numbers of hepatocytes per microscopic field in postpartum non-lactating mice resembled hepatocyte densities in livers from non-pregnant mice (Figure 4). The data indicate that hepatocyte hypertrophy also contributes to maternal liver mass expansion during pregnancy. Taken together, we conclude that pregnancy-induced hepatomegaly is a physiological event of liver growth underlying which are hepatocyte hyperplasia and hypertrophy.
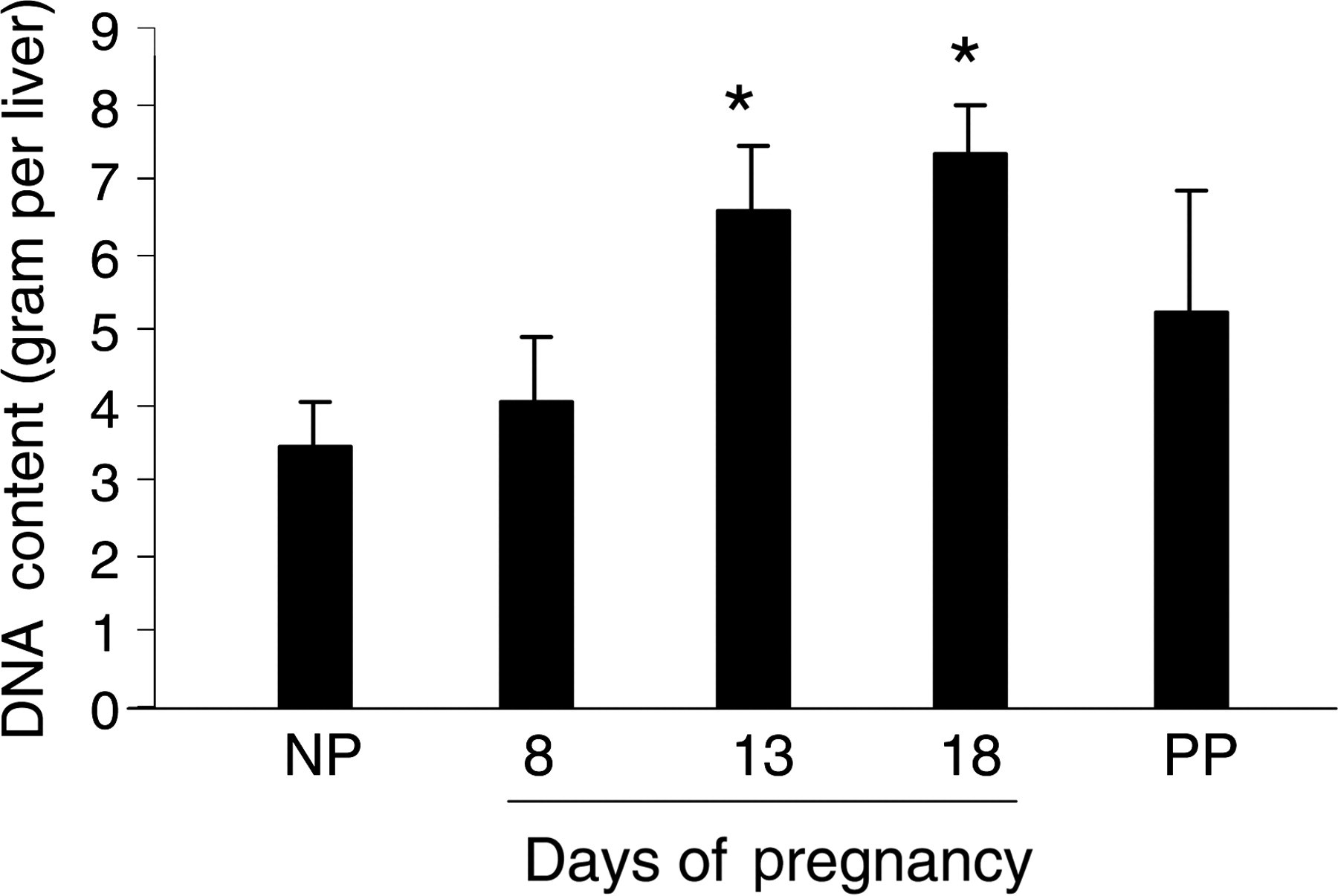
Measurement of total liver DNA content. Liver tissues collected from non-pregnant (NP), pregnant (gestation days 11, 18 and 21) and postpartum day 10 (non-lactating; PP) CD-1 mice were completely digested overnight. Total hepatic DNA content was measured using PicoGreen dsDNA Quantitation Reagent (Molecular Probes) according to the manufacturer's instructions. DNA concentrations were determined with a standard curve of fluorescence emission intensity versus DNA concentration. Data are expressed as mean ± SEM (n = 5). Asterisks indicate P < 0.05 as compared with non-pregnant mice. Note that the change of total hepatic DNA content resembled the gestational pattern of liver weight change
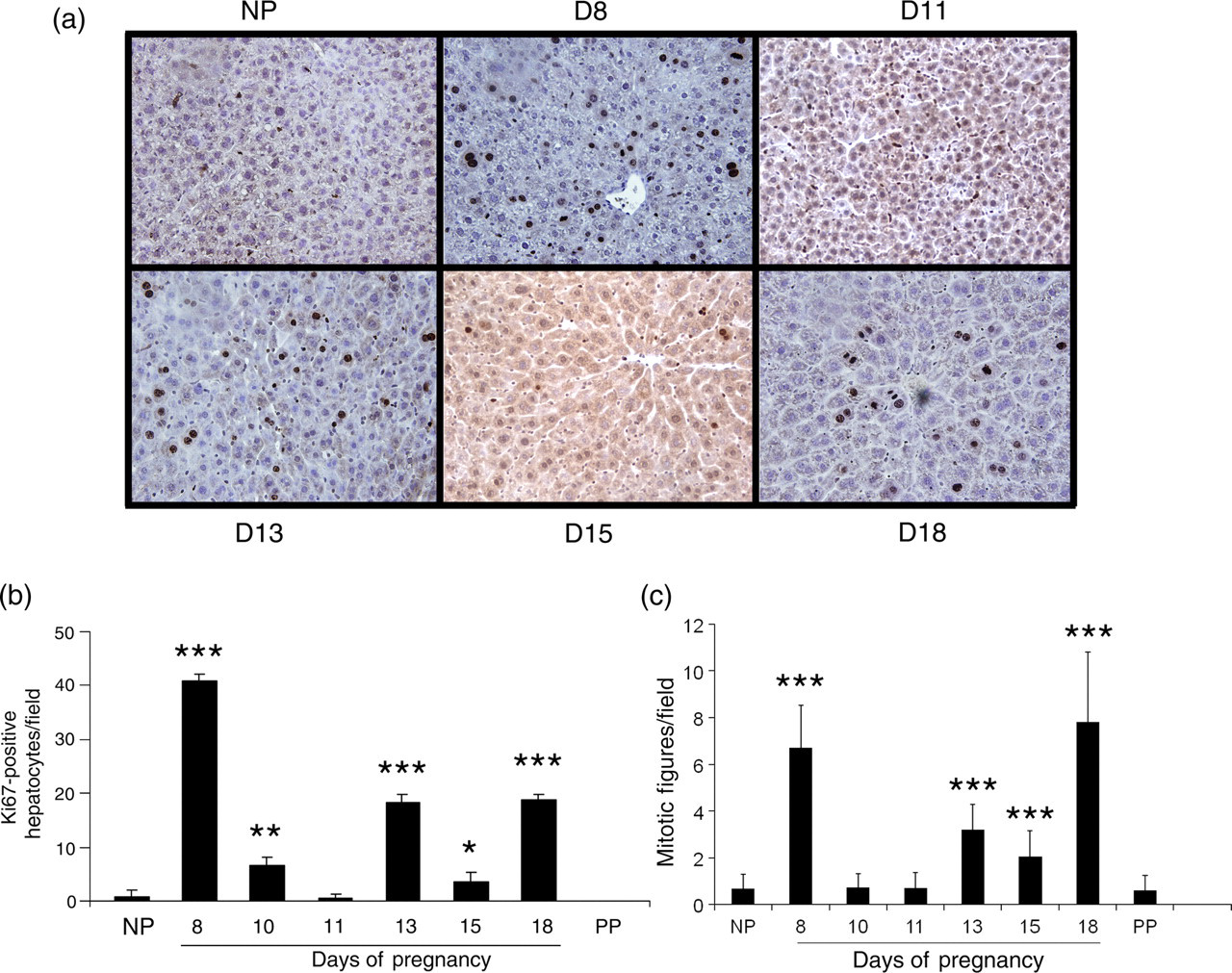
Ki-67 immunostaining in liver tissue. Liver tissues were collected from non-pregnant (NP), pregnant (gestation days 4, 8, 10, 11, 13, 15 and 18) and day 10 postpartum (non-lactating; PP) CD-1 mice, fixed in formalin and embedded in paraffin. Liver tissue sections were prepared and Ki-67 immunostaining was performed. The nuclei of Ki67-positive cells were stained dark brown. (a) Representative liver sections immunohistochemically stained for Ki-67 are shown for non-pregnant and pregnant (gestation days 8, 11, 13, 15 and 18) mice. (b) Ki67-positive hepatocytes and (c) hepatocyte mitotic figures were counted (×40 optical field) and the results are shown as mean per field ± SD (n = 3–5). Asterisks indicate the level of significance (*P < 0.05; **P < 0.01; ***P < 0.001) in comparison with non-pregnant mice. Note that hepatocyte proliferation was detected in the livers of pregnant, but not non-pregnant mice and was most prominent on gestation days 8, 13 and 18
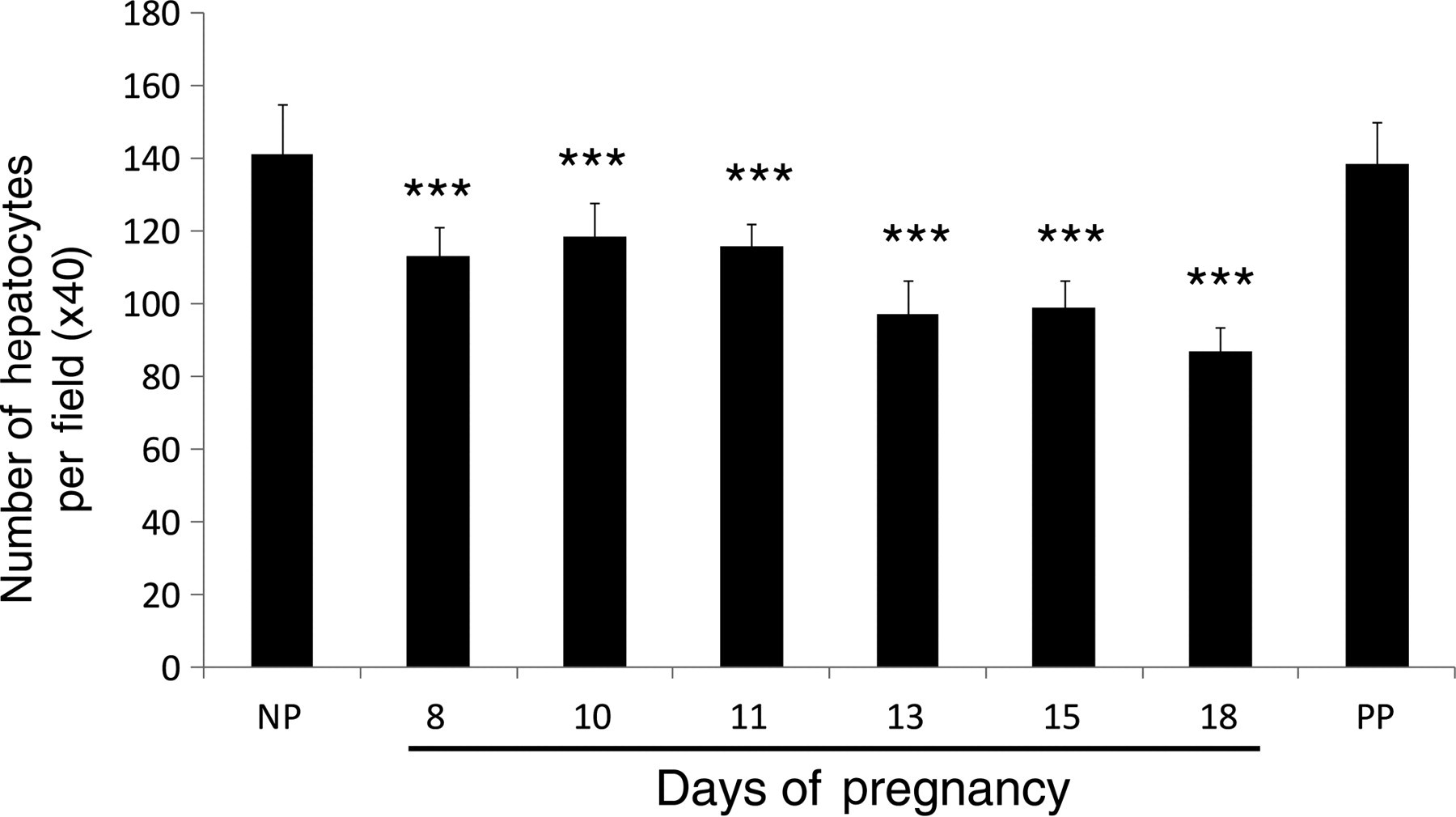
Determination of the number of hepatocytes per microscopic field. Liver tissues were collected from non-pregnant (NP), pregnant (gestation days 8, 10, 11, 13, 15 and 18) and day 10 postpartum (non-lactating; PP) CD-1 mice, fixed in formalin and embedded in paraffin. Liver tissue sections were subjected to hematoxylin and eosin staining. Hepatocytes were counted (×40 optical field) with Image-Pro Plus software (Media Cybernetics) and the results are shown as mean per field ± SD (n = 3–5). Asterisks indicate the level of significance (***P < 0.001) as compared with non-pregnant mice. Note the gestation-dependent reduction in the number of hepatocytes per field
To further evaluate pregnancy-induced hepatocyte proliferation, hepatic protein expression of several cell cycle regulators was measured. Cyclin E expression was upregulated throughout the entire course of pregnancy, whereas cyclin A2 and D1 were dynamically regulated (Figure 5). On gestation day 8 when hepatocyte proliferation was most prominent (Figure 3b), all three cell cycle regulators were activated. However, at gestation days 13 and 18 when the second and third major waves of hepatocyte proliferation occurred (Figure 3b), cyclin E and A2 expression was dramatically elevated, while cyclin D1 was downregulated. The results suggest distinct mechanisms regulating hepatocyte proliferation at different stages of maternal liver growth.
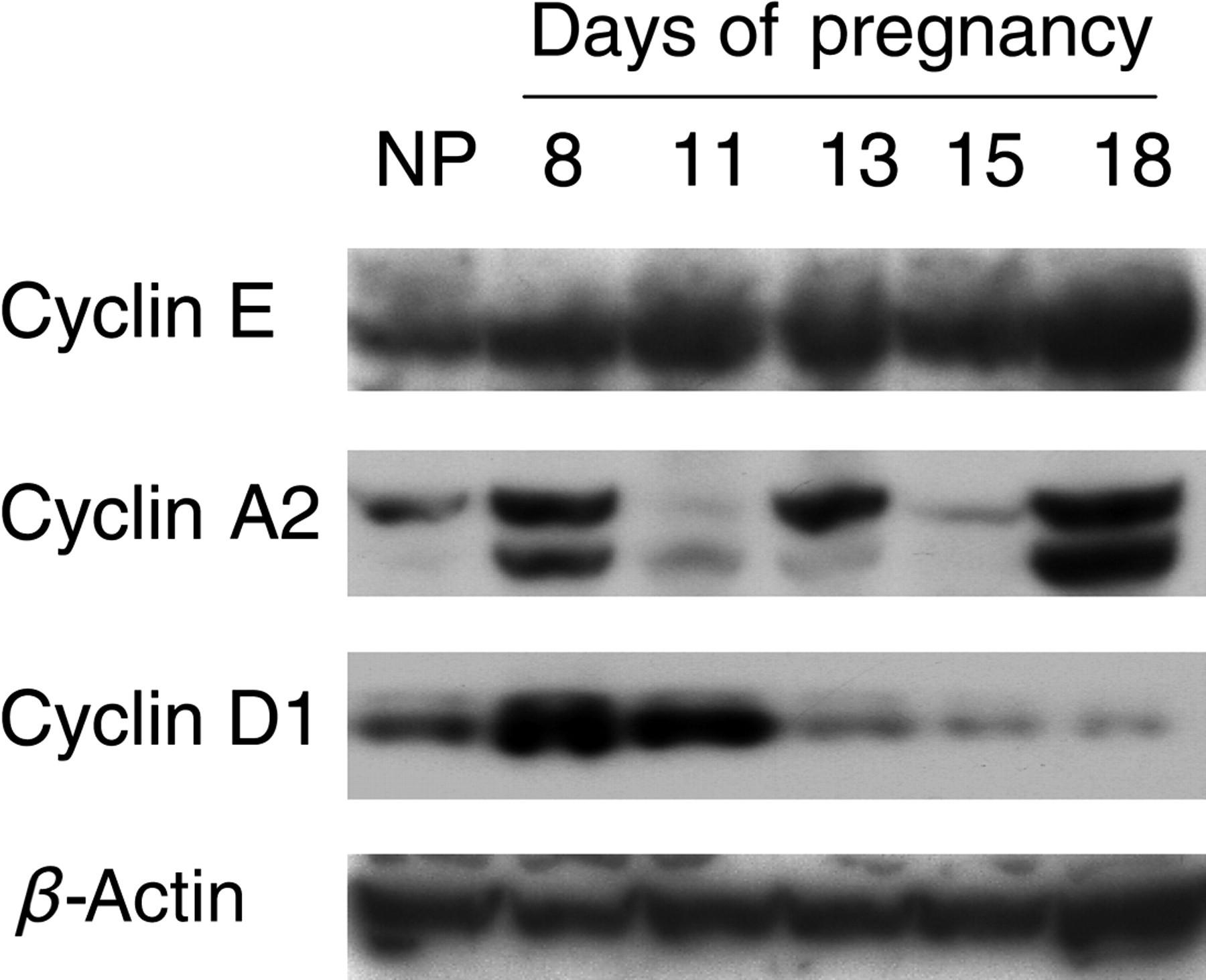
Protein expression of cell cycle regulators in the liver. Liver tissues from non-pregnant (NP) and pregnant (gestation days 8, 11, 13, 15 and 18) CD-1 mice were homogenized. Western blotting was performed on tissue homogenates using cyclin E, A2 and D1 antibodies. β-Actin protein concentrations were used as loading controls. Note the activation of cell cycle regulators by pregnancy
Pregnancy-induced liver growth is associated with a subset of genes involved in liver regeneration
To gain insights into the molecular mechanisms responsible for maternal hepatic growth response to pregnancy, the expression of several genes known to be involved in liver regeneration was evaluated in non-pregnant and pregnant mice (Figure 6). Hepatic mRNA levels of interleukin (IL)-6 and tumor necrosis factor (TNF-α), two proinflammatory factors crucial for regulating the early regenerative response of the liver, 38,39 were significantly upregulated during the course of gestation (Figures 6a and b). An increase in hepatic expression of c-Jun, a transcription factor regulating hepatocyte proliferation in response to liver mass loss, 40 was evident during the first half of pregnancy (Figure 6c). Hepatic expression of IL-1β, a factor that curbs hepatocyte DNA synthesis during liver re-growth, 41 was elevated during pregnancy and following parturition (Figure 6d). Collectively, the dynamic changes in expression of this subset of genes implicated as regulators of injury-induced liver regeneration suggest that they may also participate in regulating maternal hepatic growth responses to pregnancy.
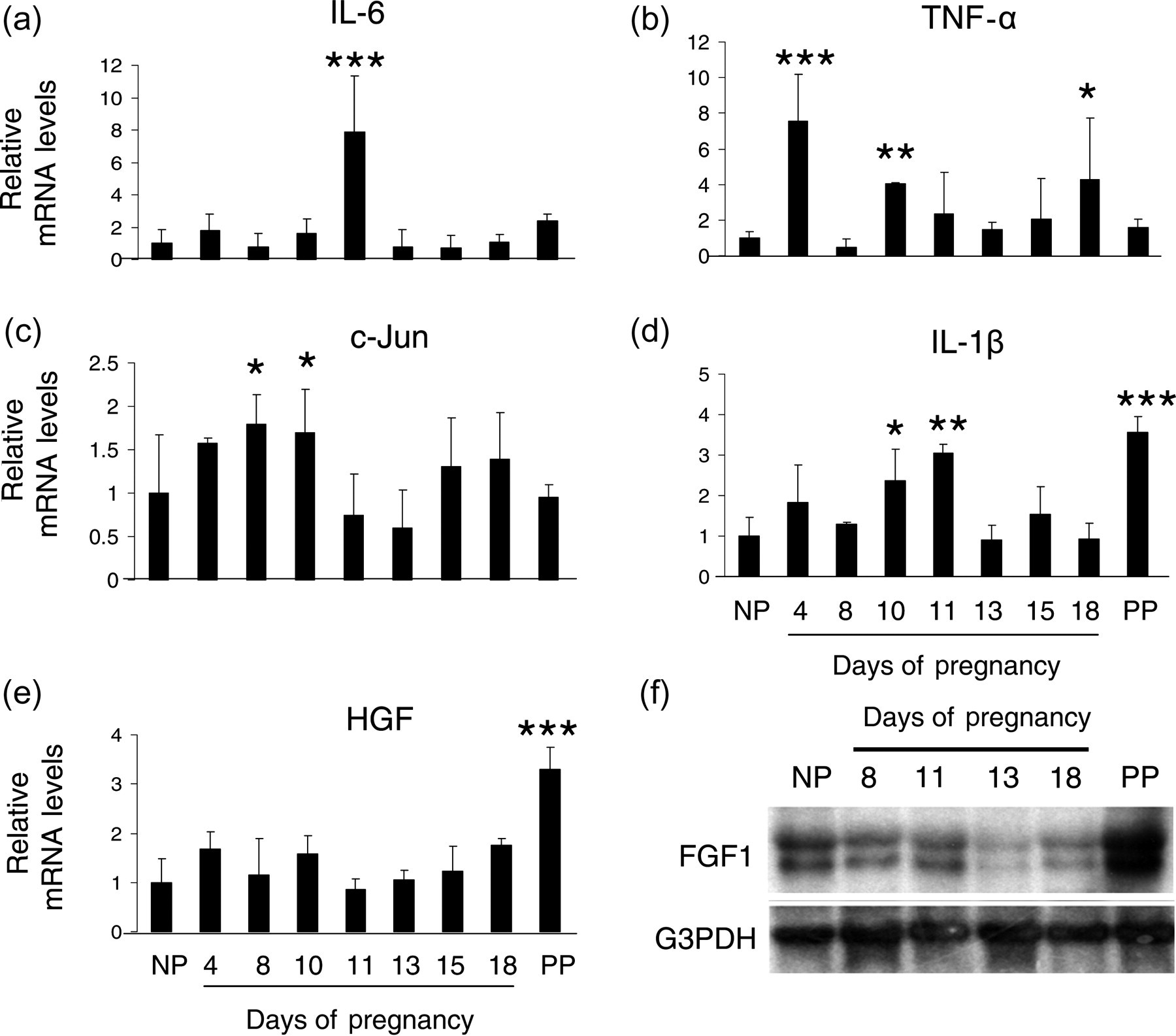
Hepatic expression of genes involved in liver regeneration during pregnancy. Total RNA was prepared from liver tissues of non-pregnant (NP), pregnant (gestation days 4, 8, 10, 11, 13, 15 and 18) and day 10 postpartum (non-lactating; PP) CD-1 mice, respectively. Hepatic mRNA levels of interleukin (IL)-6 (a), tumor necrosis factor (TNF) α (b), c-Jun (c), IL-1β (d) and hepatocyte growth factor (HGF) (e) were quantified by realtime polymerase chain reaction and are expressed as mean of fold changes compared with non-pregnant controls ± SD (n = 3 for each group). Asterisks indicate the level of significance (*P < 0.05; **P < 0.01; ***P < 0.001) in comparison with non-pregnant mice. Hepatic gene expression of fibroblast growth factor 1 (FGF1) (f) was analyzed by Northern blot. Twenty micrograms of total RNA was loaded on 1% agarose gels. A cDNA probe for FGF1 was labeled with alpha-[32P]dCTP. G3PDH served as an internal control demonstrating equal loading and RNA integrity. Note that the genes indicated exhibited differential expression patterns during pregnancy
Hepatic transcript levels of hepatocyte growth factor (HGF) and FGF1, two potent growth factors stimulating cell proliferation during liver regeneration, 42,43 were either not significantly affected by pregnancy (Figure 6e) or even decreased with the progression of pregnancy (Figure 6f). Increases of hepatic expression of both HGF and FGF1 genes were seen postpartum (Figures 6e and f). These observations indicate that pregnancy-induced maternal hepatic growth is probably regulated by factors other than hepatic HGF and FGF1.
Deficiency of TNFR1 has no significant impact on pregnancy-dependent liver growth
TNFR1-null mice display disruptions in liver regeneration. 44–46 This prompted us to examine whether TNFR1 participates in the regulation of maternal liver growth induced by pregnancy. Maternal livers were collected and weighed from non-pregnant and pregnant (gestation days 13 and 18) wild-type and TNFR1-null C57BL/6 mice. Both wild-type and TNFR1 null mice exhibited pregnancy-dependent increases in liver growth (Figure 7). The results indicate that lack of TNFR1 signaling does not significantly affect pregnancy-dependent liver growth.
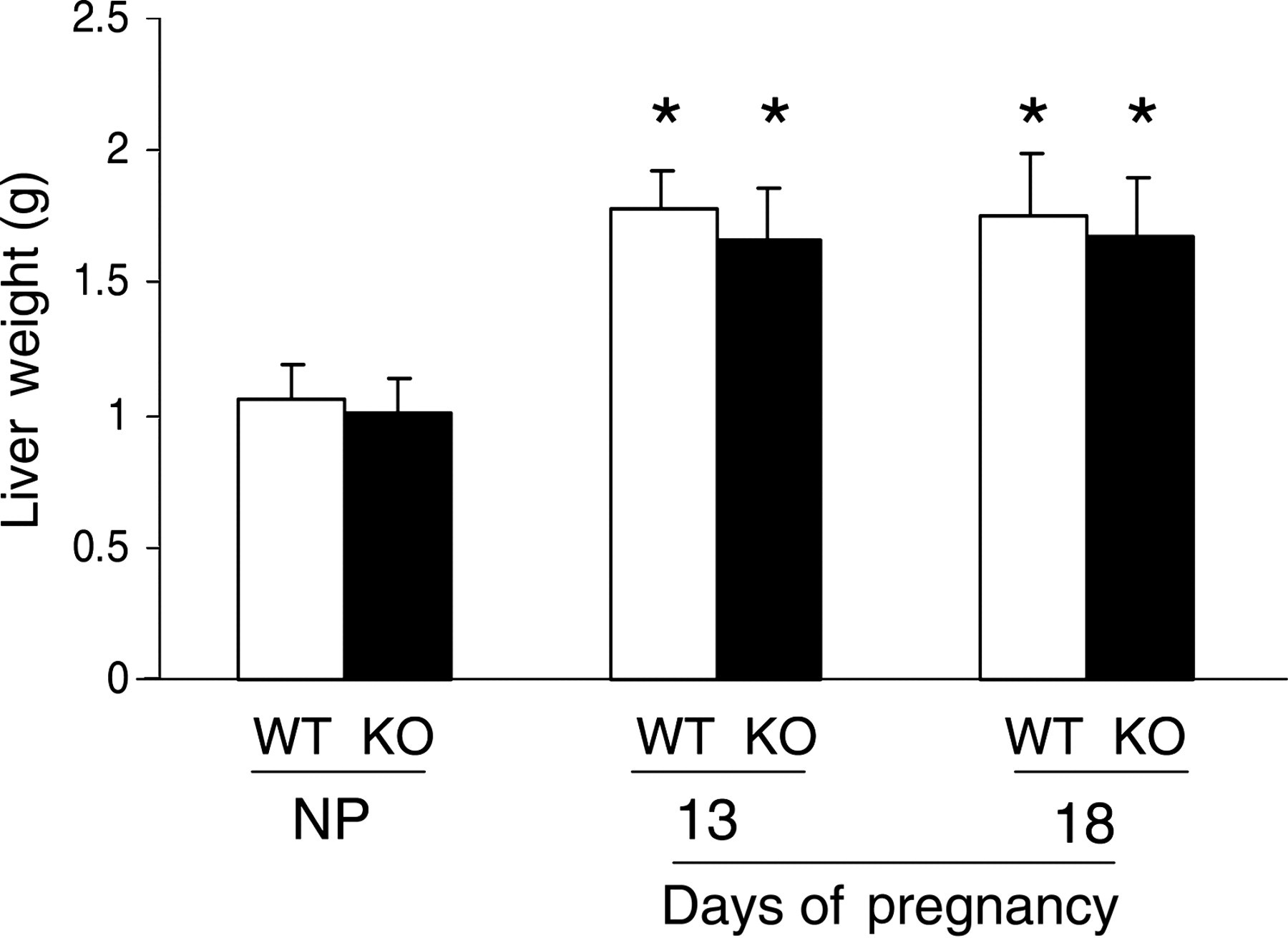
Maternal liver weight responses to pregnancy in wild-type (WT) and TNFR1 (tumor necrosis factor receptor 1)-null (KO) mice. Livers were isolated and weighed from non-pregnant (NP) and gestation days 13 and 18 pregnant WT and KO mice. Data are expressed as mean of weight (g) ± SEM. Asterisks indicate P < 0.05 (n = 5) as compared with non-pregnant mice. Note the lack of a significant impact of TNFR1 signaling on pregnancy-induced liver growth
Hepatic pregnane X receptor and constitutive androstane receptor are not activated during pregnancy
Pregnane X receptor (PXR) and constitutive androstane receptor (CAR) are two primary xenobiotic receptors that regulate the expression of genes involved in phase I, phase II and phase III metabolism of xenobiotics and endobiotics. 47,48 Notably, upon activation, both PXR and CAR induce hepatocyte proliferation, resulting in hepatomegaly. 49,50 In addition, PXR participates in regulating liver regeneration. 51 Thus, determination of the expression and functional status of these two nuclear receptors in livers from pregnant mice provides insight into their participation in pregnancy-induced hepatomegaly. Therefore, transcript levels of PXR, Cyp3a11 (a prototypical PXR target gene), CAR and Cyp2b10 (a prototypical CAR target gene) were measured by qRT-PCR during the course of gestation (Figure 8). Hepatic PXR (Figure 8a), Cyp3a11 (Figure 8b), CAR (Figure 8c) and Cyp2b10 (Figure 8d) mRNAs did not exhibit significant increases during pregnancy. In contrast, significant pregnancy-dependent decreases of hepatic Cyp3a11 (Figure 8b), CAR (Figure 8c) and Cyp2b10 (Figure 8d) transcript levels were observed at midgestation (gestation day 10 and/or 11). Thus hepatic PXR and CAR are not activated by pregnancy, suggesting that these two nuclear receptors are unlikely to be the mediators of maternal hepatic growth response to pregnancy.
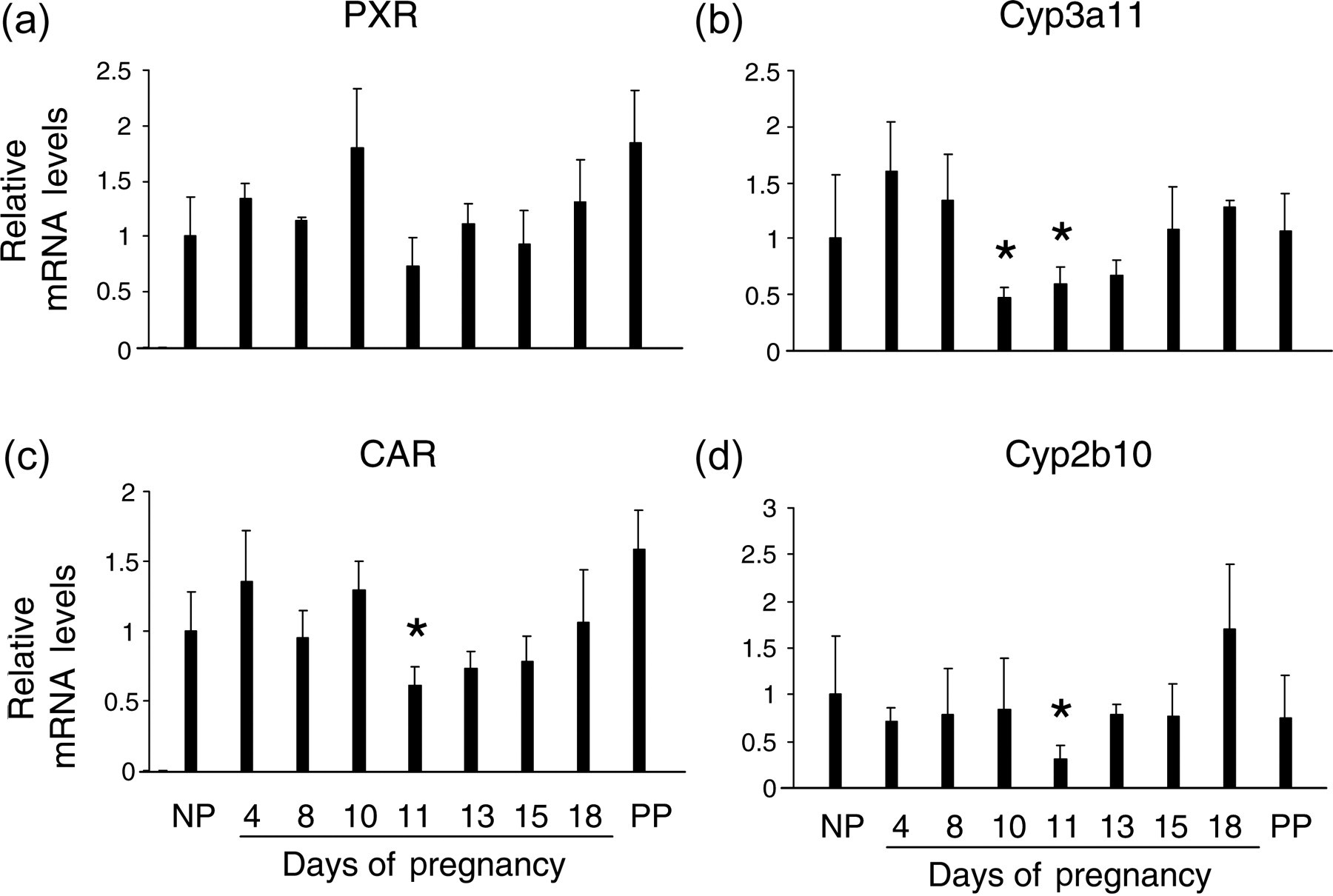
Hepatic gene expression of pregnane X receptor (PXR), Cyp3a11, constitutive androstane receptor (CAR) and Cyp2b10 during pregnancy. Total RNA was prepared from liver tissues of non-pregnant (NP), pregnant (gestation days 4, 8, 11, 13, 15 and 18) and day 10 postpartum (non-lactating; PP) mice, respectively. Hepatic mRNA levels for PXR (a), Cyp3a11 (b), CAR (c) and Cyp2b10 (d) were measured by quantitative realtime reverse transcriptase polymerase chain reaction and are expressed as means of fold changes compared with non-pregnant controls ± SD (n = 3 for each group). Asterisks indicate P < 0.05. Note the absence of a pregnancy-dependent increase in hepatic mRNA levels for each of these genes
A subset of signaling molecules regulating liver regeneration are activated in maternal liver during pregnancy
Both pregnancy-induced liver growth and liver regeneration are driven by hepatocyte proliferation (current study and Michalopoulos 42 ). The functional states of several signaling molecules known to regulate liver regeneration were investigated in non-pregnant and pregnant mice. Hepatic protein levels of phosphorylated STAT3 were markedly increased throughout gestation, indicating robust activation of the molecule, while gestation-dependent increases in hepatic content of total STAT3 protein were much less striking (Figure 9). Hepatic non-phosphorylated β-catenin exhibited an increase especially during the second half of pregnancy (Figure 9), in parallel with the maternal liver growth pattern (Figure 1), whereas total β-catenin did not show marked changes (Figure 9). The results demonstrate that pregnancy activates STAT3 and β-catenin-mediated signaling pathways in the maternal liver. Hepatic phosphorylated EGFR showed increases from gestation day 8 to 15, followed by a decrease prior to parturition (gestation day 18), without changes in total EGFR throughout pregnancy (Figure 9). The data indicate that hepatic EGFR signaling is dynamically regulated by pregnancy. Surprisingly, both phosphorylated and total c-Met, a receptor mediating potent mitogenic effect of HGF during liver regeneration, 39,42 did not exhibit increased hepatic expression during pregnancy (Figure 9). The finding indicates that c-Met may not be a major player in mediating maternal hepatic growth response to pregnancy.
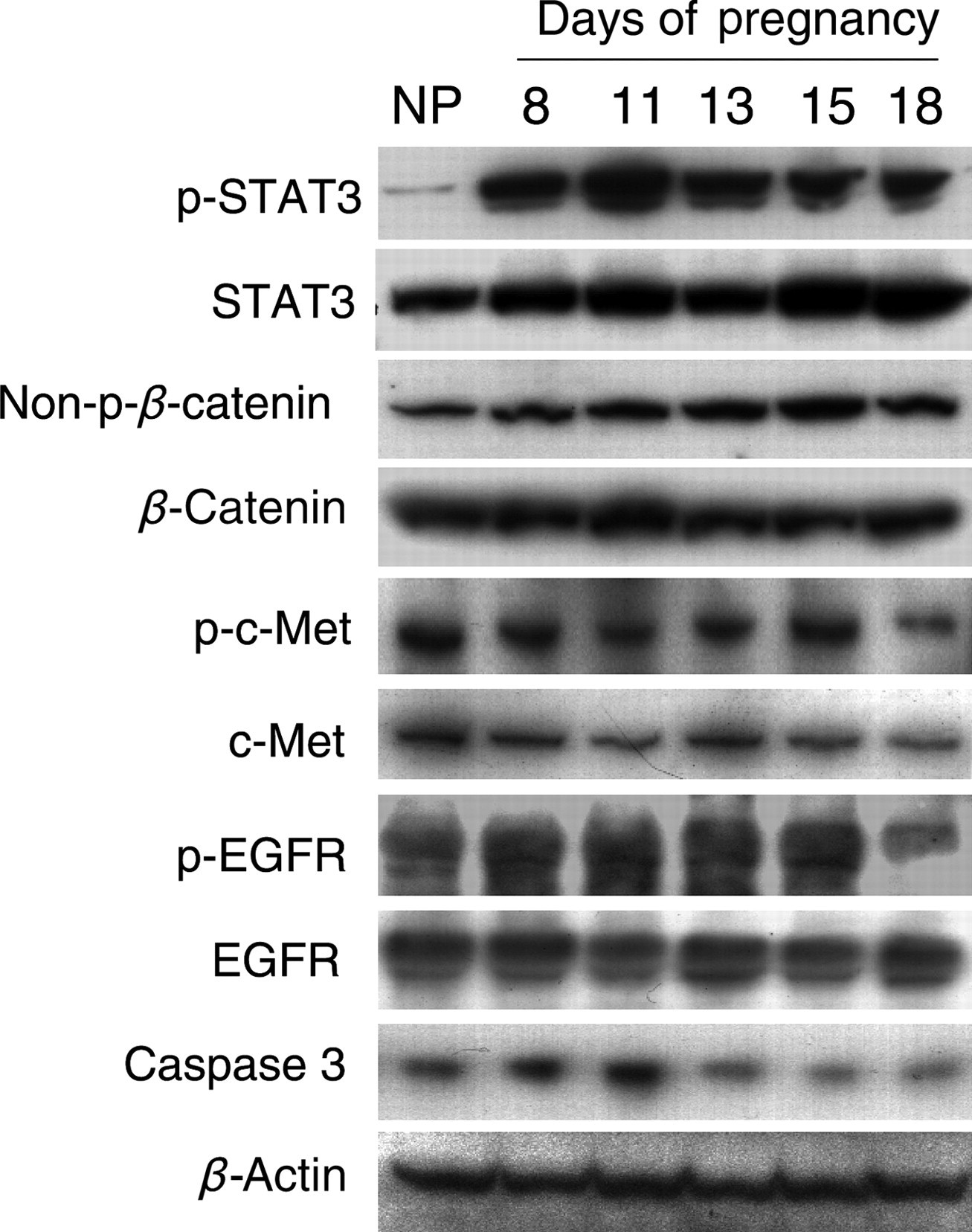
Protein expression and functional states of a subset of signaling molecules regulating liver regeneration during pregnancy. Liver tissues from non-pregnant (NP) and pregnant (gestation days 8, 11, 13, 15 and 18) CD-1 mice were homogenized. Western blotting was performed on tissue homogenates using antibodies against the proteins indicated. β-Actin protein levels were used as loading controls. Note the activation of STAT3 and β-catenin by pregnancy. EGFR, epidermal growth factor receptor; STAT3, signal transducer and activator of transcription 3
Additionally, liver regeneration is accompanied by hepatic apoptosis. 39,42 To determine whether this event also occurs during maternal liver growth, hepatic caspase 3 protein was examined in non-pregnant and pregnant mice. Unexpectedly, although total caspase 3 protein level showed an increase during the first half of pregnancy (Figure 9), cleaved caspase 3 was not detected at all the time points examined (data not shown). The data suggest that apoptosis may not be a major event during pregnancy-induced maternal liver growth.
Discussion
The current study revealed that significant liver growth takes place during pregnancy in the mouse. In normal conditions, the liver is a largely quiescent organ despite its large metabolic load, maintaining a steady mass, which is controlled by a delicate balance between cell gain and cell loss. 52 A very small proportion of hepatocytes (<0.01%), the major structural and functional cells in the liver, undergo mitosis at any given time. 38,52 However, a high capacity of hepatocyte replication and hepatic regeneration can be elicited with an appropriate stimulus. Pathological stimuli, acute or chronic liver injuries induced by various insults, including toxins, drug toxicity, viral infection or surgical resection, can trigger the hepatic regenerative response manifested by hepatocyte proliferation to replace the damaged tissue. 53 This phenomenon of hepatic regeneration is also considered as compensatory growth of the liver. 39 Chemical stimuli, including certain xenobiotics (e.g. barbiturates and pregnenolone 16α-carbonitrile), which are capable of activating specific nuclear receptors, such as PXR and CAR, induce hepatocyte proliferation and liver enlargement (hepatomegaly), increasing the capacity of the liver for xenobiotic detoxification. 54,55 This phenomenon represents a hepatic adaptive response to alleviate xenobiotic stress. 49 In this report, pregnancy was identified as a physiological stimulus capable of stimulating maternal liver growth to meet the metabolic needs of the growing placenta and fetus.
Our report demonstrates that maternal liver growth during pregnancy is driven by hepatocyte hyperplasia and hypertrophy. Several waves of hepatocyte proliferation, manifested by increases in the numbers of Ki67-positive hepatocytes and mitotic figures and in maternal hepatic DNA content (Figure 3), were evident as pregnancy progresses. The most prominent waves of hepatocyte proliferation took place on gestation days 8, 13 and 15 (Figure 3). Hepatocyte hypertrophy was concomitant with maternal liver mass expansion, especially during the second half of pregnancy (Figure 4). It is noteworthy that, by examining non-pregnant and gestation day 18 livers, Williamson's group observed gestation-dependent increases in the numbers of Ki67-positive heaptocytes and hepatocyte hypertrophy, whereas mitotic figures were not seen. 35 Thus, it was concluded that pregnancy-induced liver growth is mainly a result of hepatocyte hypertrophy caused by the cell cycle arrest before mitosis. However, by investigating non-pregnant and gestation days 8, 10, 11, 13, 15 and 18 livers, we saw gestation-dependent increases in both the numbers of Ki67-positive hepatocytes and mitotic figures at several gestation days, in addition to hepatocyte hypertrophy (Figure 3). Here, our conclusion is that hepatocyte proliferation and size increase are the driving forces for maternal liver growth during pregnancy. The discrepancy in the conclusion between our group and Williamson's group might be caused by the number of time points examined during the course of gestation and/or possible differences of mouse strains in maternal hepatic response to pregnancy.
We noticed that, during the first half of pregnancy, although maternal liver mass was increased as the first wave of hepatocyte proliferation took place, statistical significance between non-pregnant and pregnant livers in weight was not achieved (Figure 1). Robust maternal liver mass expansion occurred during the second half of pregnancy as the continuation of hepatocyte proliferation and cell size increased (Figures 1a, 3b and 4). Likewise, in the mouse model of 70% partial hepatectomy, hepatocyte proliferation peaked at 36 h after surgery, whereas significant liver mass increases were not observed until several days following the hepatocyte proliferation peak. 51,56 Thus, pregnancy-dependent liver mass expansion is an outcome of a complicated biological process associated with an initial increase in cell proliferation and subsequent hepatocyte hypertrophy and tissue remodeling.
Our studies have identified three major waves of maternal hepatocyte proliferation during the course of gestation, which may be regulated by different mechanisms. On gestation day 8 when the first wave of maternal hepatocyte proliferation occurred (Figure 3), cyclins E, A2 and D1 were all upregulated (Figure 5). In contrast, on gestation days 13 and 18 when the second and third major waves of hepatocyte proliferation took place (Figure 3), cyclins E and A2 were activated without participation of cyclin D1 in the cell cycle (Figure 5). It has been demonstrated that cyclin D1 is not essential for hepatocyte proliferation induced by mitogenic stimuli and the overexpression of cyclin E may compensate for the lack of cyclin D1. 57 Therefore, it may not be a surprise that a disconnection between maternal hepatocyte proliferation and cyclin D1 was observed. Notably, on gestation day 11, activation of cyclin E and D1 was not accompanied by proliferation of hepatocytes (Figures 3 and 5), but instead non-parenchymal cells (Figure 3a). Our findings implicate stage- and cell type-dependent regulation of the cell cycle by different mitogens or signaling pathways in the maternal liver during the course of gestation.
Our studies indicate that pregnancy-induced liver growth and liver regeneration possess commonalities and differences in molecular mechanisms. Liver regeneration is a well-orchestrated process associated with signaling cascades involving growth factors, cytokines and matrix remodeling. 42 Several genes known to participate in the regulation of liver regeneration, including IL-6, TNF-α, c-Jun and IL-1β, were upregulated in a stage-dependent manner during the course of pregnancy (Figure 6). The findings suggest that these genes may also be involved in the regulation of maternal hepatic growth response to pregnancy. However, unlike liver regeneration, maternal hepatocytes undergo several waves of proliferation and concomitantly increase their sizes in response to increasing metabolic demands from maternal, placental and fetal compartments. At this juncture, it remains an open question how these genes are integrated into the regulatory machinery responsible for maternal liver growth. Remarkably, STAT3, β-catenin and EGFR are activated in maternal liver by pregnancy (Figure 9). STAT3 is required for hepatocyte DNA synthesis and survival in the acute phase of liver regeneration. 58 The essential roles of β-catenin, an integral component of the Wnt signaling pathway, in liver growth, development and regeneration have been extensively studied. 59 EGFR mediates signaling of a number of growth factors, such as EGF, transforming growth factor (TGF)-α and heparin-binding EGF, that drive cell cycle progression during liver regeneration. 39 In our studies, increases in EGFR phosphorylation were seen in maternal livers at most of the time points examined (Figure 9). Thus, STAT3, β-catenin and EGFR, together with other molecules mentioned above, may represent some mechanisms shared by pregnancy-induced liver growth and liver regeneration.
Notably, estrogen receptor signaling should be considered in further mechanistic studies in understanding maternal liver growth during pregnancy. The placenta synthesizes and elaborates abundant estrogens required for maintaining pregnancy. In vitro studies showed that, as co-mitogens for hepatocytes, estrogens enhance the mitogenic effects of EGF, whereas they inhibit the effects of the mito-inhibitor TGF-β1, on hepatocytes. 60–63 In vivo studies demonstrated that estrogen induces transient hepatocyte proliferation and liver growth. 64–66 Thus, it is presumable that the increase in estrogens driven from the placenta enhances the effects of EGF continually supplied to the liver through the portal circulation (Brunner's glands of the duodenum) and simultaneously inhibits the effects of TGF-β1, leading to maternal liver growth. Moreover, estrogen receptor is involved in regulating liver regeneration. 66–69 Therefore, estrogen receptor signaling may also be a mechanism shared by both pregnancy-induced liver growth and injury-induced liver regeneration.
On the other hand, our investigation provides several lines of evidence to support our notion that the regulatory mechanisms underlying maternal hepatic growth response to pregnancy are distinct from mechanisms controlling liver regeneration. HGF, the most potent direct mitogen for hepatocytes identified so far, has been demonstrated as an initiator of liver regeneration and signaling by HGF appears as the most irreplaceable contributor to liver regeneration. 42 Targeted genetic elimination of HGF receptor from the liver leads to a compromised regenerative response. 70,71 Surprisingly, maternal hepatic HGF mRNA expression and the functional status of its receptor c-Met were not significantly affected by pregnancy (Figures 6e and 9). As a potent mitogen for hepatocytes, FGF1 is also considered to be a modulator of liver regeneration. 43 However, hepatic FGF1 expression declines as pregnancy advances (Figure 6f). TNFR1-mediated TNF signaling is critical for the initiation of liver regeneration induced by both partial hepatectomy and exposure to toxic chemicals. TNFR1 deficiency results in severe suppression of cell proliferation during liver regeneration. 44–46 In contrast, genetic deletion of TNFR1 did not significantly alter the liver growth pattern during pregnancy (Figure 7). Moreover, PXR and CAR, which participate in regulating liver regeneration, 51,72 were not activated by pregnancy (Figure 8). These two nuclear receptors can be activated by naturally occurring steroid hormones, such as pregnenolone and progesterone, and other endobiotics, such as bilirubin and bile acids. 47,55,73 It is surprising that, in an environment with high concentrations of steroid hormones required for maintaining pregnancy, these two nuclear receptors are not activated. We conclude that pregnancy-induced maternal liver growth and injury-induced liver regeneration exhibit similarities and differences in their regulation.
A recent report demonstrated that the aged maternal liver regenerates via hepatocyte hypertrophy after partial hepatectomy during pregnancy. 74 The finding indicates potent effects of pregnancy on maternal liver re-growth after massive mass loss. Notably, other recent reports highlighted the role of placental hormones, such as prolactin and placental lactogens, in mediating maternal pancreatic growth response to pregnancy. 3,4 Whether these hormones take part in regulating maternal liver growth during pregnancy is under investigation.
In summary, the current study revealed that the liver of the mouse adapts to the demands of pregnancy via a significant growth response induced by hepatocyte hyperplasia and hypertrophy. Gestational-dependent maternal hepatic growth occurs post-implantation and coincides with the growth and development of the placenta and fetus. The regulatory machineries controlling pregnancy (physiological)-induced liver growth and injury (pathological)-induced liver re-growth possess commonalities and differences. Our study provides insight into a novel mouse model for investigating the regulation of hepatocyte proliferation and liver growth. Further investigation needs to be performed to evaluate the maternal hepatic functional changes induced by pregnancy and the molecular and cellular mechanisms governing the hepatic adaptive response to pregnancy. This will not only further our understanding of liver physiology during pregnancy but also provide insights into mechanisms underlying pregnancy-associated liver diseases, as well as predict the influence of pregnancy on drug metabolism.
Footnotes
ACKNOWLEDGEMENTS
We thank Drs Kathy F Roby and Paul F Terranova of the University of Kansas Medical Center for providing the TNFR1 null mice. This work was supported by grants from the Andrew Mellon Foundation, the National Institute of Child Health and Human Development (HD39878, HD48861) and the National Institute of Diabetes and Digestive and Kidney Diseases (7RO1DK07596).