
Abstract
Ovarian cancer is the leading cause of death from gynecological malignancies. Although initial therapeutic modalities are successful, 65% of these women relapse with only palliative treatments available thereafter. Endogenous opioids repress the proliferation of human ovarian cancer cells in vitro, and do so in a receptor-mediated manner. The present study examined whether modulation of opioid systems by the opioid antagonist naltrexone (NTX), alone or in combination with standard of care therapies (taxol/paclitaxel, cisplatin), alters human ovarian cancer cell proliferation in tissue culture and tumor progression in mice. Administration of NTX for six hours every two days, but not continuously, reduced DNA synthesis and cell replication from vehicle-treated controls in tissue culture. Moreover, brief exposure to NTX in combination with taxol or cisplatin had an enhanced anticancer action. Mice with established ovarian tumors and treated with a low dosage of NTX (LDN), which invokes a short period of opioid receptor blockade, repressed tumor progression in a non-toxic fashion by reducing DNA synthesis and angiogenesis but not altering cell survival. The combination of LDN with cisplatin, but not taxol, resulted in an additive inhibitory effect on tumorigenesis with enhanced depression of DNA synthesis and angiogenesis. LDN combined with cisplatin alleviated the toxicity (e.g. weight loss) associated with cisplatin. LDN treatment upregulated the expression of the opioid growth factor (OGF, chemical term ([Met5]-enkephalin) and its receptor, OGFr. Previous tissue culture studies have reported that OGF is the only opioid peptide with antiproliferative activity on ovarian cancer cells, with OGF action mediated by OGFr. Thus, the common denominator of intermittent opioid receptor blockade by short-term NTX or LDN on ovarian cancer proliferation and tumorigenesis recorded herein appears to be related to the OGF–OGFr axis. These preclinical data may offer a non-toxic and efficacious pathway-related treatment that can benefit patients with ovarian cancer.
Keywords
Introduction
Ovarian cancer is the fifth leading cause of cancer mortality among women in the USA and the major cause of death from gynecological malignancies. 1,2 More than 75% of women are diagnosed with ovarian cancer in advanced stages. Ninety percent of these neoplasias are epithelial in origin. 3 Although the initial clinical response to cytoreductive surgery and adjuvant chemotherapy is excellent, nearly 65% of advanced-staged patients relapse within two years and, upon recurrence, all subsequent treatments are palliative 1,4 The cellular and molecular events involved in the pathogenesis of this deadly neoplasm need to be defined, as major improvements in the prognosis and treatment of ovarian cancer patients will necessitate novel therapies that target biological pathways. 1
Endogenous opioids and opioid receptors have been shown to have tonically active growth regulatory properties in neoplasia, including human ovarian cancer. 5,6 Opioid antagonist modulation of endogenous opioid systems has been used to decipher the function of opioid peptide–opioid receptor interactions in a number of biological processes and diseases including cancer. 7–12 Naltrexone (NTX) is a general opioid receptor antagonist 13 that is devoid of intrinsic activity 14–16 and blocks endogenous opioids from opioid receptors. This opioid receptor blockade leads to an upregulation in the production of endogenous opioid peptides and opioid receptors. 17–20 Continuous opioid receptor blockade, achieved using a daily high dose of NTX (e.g. 10 mg/kg in mice) or administration of a low dose of naltrexone (LDN) (e.g. 0.1 mg/kg in mice) multiple times each day, does not permit the interfacing of the upregulated opioids and receptors. 7,21,22 Such a constant opioid receptor blockade results in repercussions such as an acceleration in DNA synthesis and tumor progression. 8,21,22 However, pharmacokinetic, 23 nociceptive 7,21 and functional 7,10,20,24 studies have shown that blockade of opioid peptides from opioid receptors for a short period each day (4–6 h), using a daily administration of LDN, provides an 18–20 h window wherein the elevated levels of endogenous opioids and opioid receptors can interact to elicit a response (e.g. depression of DNA synthesis, inhibition of tumorigenesis). 8,21,22,24–27
One particular endogenous opioid peptide–opioid receptor system involved in growth modulation by NTX has been demonstrated to upregulate is the opioid growth factor (OGF) and its receptor, OGFr. 9,28 OGF, chemically termed [Met5]-enkephalin, is a constitutively expressed native opioid peptide that is autocrine produced and secreted. 29 OGF interacts with OGFr (a non-classical opioid receptor) to delay the G1/S phase of the cell cycle by modulating cyclin-dependent kinase inhibitory (CKI) pathways, and inhibits cell proliferation in normal and neoplastic cells, including ovarian cancer. 5,6,29–32 The OGF–OGFr axis has been shown to be present in human ovarian cancer, with OGFr RNA, protein and binding activity documented in these cells in vitro, 5 and OGF detected by radioimmunoassay in surgical samples taken from human ovarian neoplasms. 33 Studies in tissue culture have documented that OGF is the only endogenous opioid peptide which regulates ovarian cancer cell proliferation, 5 and that the inhibitory action of OGF is mediated by OGFr. 5,6 An increase in OGF–OGFr activity in human ovarian cancer cells in tissue culture by the addition of exogenous OGF has been reported to markedly suppress cell proliferation in a non-toxic manner by targeting the CKI pathways. 5,6 Moreover, continuous intervention of opioid peptide–opioid receptor interaction with the opioid antagonist NTX accelerates cell proliferation. 5,6
Given that the OGF–OGFr axis is present and functions in an inhibitory manner in human ovarian cancer cells in tissue culture, 5,6 and that persistent opioid receptor blockade by NTX increases DNA synthesis and cell number, 5,6 this raises the question of whether modulation of the OGF–OGFr system by intermittent opioid receptor blockade (e.g. LDN) can alter the progression of this gynecological cancer. In addition, this study inquired as to whether LDN could be combined with either of two standard of care therapies, taxol (paclitaxel) or cisplatin, to provide an added inhibitory effect on ovarian tumorigenesis. To initially address these questions, we have developed a tissue culture model that exposes ovarian cancer cells to NTX for a short period of time, resulting in suppression of DNA synthesis and cell proliferation. This in vitro model using short-term NTX exposure has compelling parallels with LDN in vivo, and allowed direct examination of the repercussions of combining a brief duration of opioid receptor blockade with taxol or cisplatin on ovarian cancer without confounding systemic influences. To gain the full perspective of the biological significance of modulating the OGF–OGFr axis with LDN, we evaluated the effects of LDN alone and in combination with chemotherapy in mice with xenografts of established human ovarian cancer.
Material and methods
Cell culture
The human ovarian cancer cell line SKOV-3 34 was obtained from The American Type Culture Collection (Manassas,VA, USA). Cells were grown in a humidified atmosphere of 5% CO2/95% air at 37°C in RPMI medium supplemented with 1.2% sodium bicarbonate, 10% fetal calf serum and antibiotics (5000 U/mL penicillin, 5 μg/mL streptomycin and 10 mg/mL neomycin).
Growth assays
Cells were plated and counted 24 h later (time 0) to determine seeding efficiency. Cultures were treated with NTX (10−5 mol/L), taxol (10−9 or 10−10 mol/L), cisplatin (0.01 or 0.001 μg/mL), NTX (10−5 mol/L) and taxol (10−9 or 10−10 mol/L), NTX (10−5 mol/L) and cisplatin (0.01 or 0.001 μg/mL), or an equivalent volume of sterile water. At the end of six hours, the media containing compound(s) was removed and replaced with media either lacking NTX (short-term NTX treatment) or containing NTX (continuous NTX treatment), taxol or cisplatin. Media and compounds were replaced on a daily basis except for the short-term NTX group, where treatment was administered every 48 h. In the reversal studies, some cultures had media replaced without compound at 48 h. Taxol was dissolved in dimethyl sulfoxide (DMSO) (10−2 mol/L) and further diluted in sterile water; all other compounds were prepared in sterile water, and dilutions represent final concentrations. An equivalent volume of vehicle was added to control (Co) wells. Cells were harvested at designated times, stained with trypan blue and counted with a hemacytometer. At least two aliquots/well and two wells/treatment/timepoint were sampled.
Animals, tumor cell implantation and tumor growth
Four-week-old athymic nu/nu female mice, purchased from the Charles River Laboratory (Wilmington, MA, USA), were housed in pathogen-free isolator ventilated cages in a temperature-controlled room (22–25°C) with a 12–12 h light/dark cycle (lights on 07:00–19:00). A sterile rodent diet (Harlan Teklad, Fredrick, MD, USA) and water were available ad libitum. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of The Pennsylvania State University College of Medicine, and conformed to the guidelines established by the NIH. Following a 48-h acclimation period, unanaesthetized mice were injected subcutaneously with SKOV-3 cells (4 × 106/mouse) into the right scapula region. Mice were weighed three times/week, observed daily for initial appearance of tumors and tumors were measured three times/week using a vernier caliper. Volume was calculated using the formula l × w 2 × π/6, where length (l) is the longest dimension and width (w) is the dimension perpendicular to the length. 35
Drug treatment
Beginning on the day tumors became visible (day 0), six groups of mice (n = 10) were randomly assigned to receive intraperitoneal injections of LDN (0.1 mg/kg, daily), taxol (3 mg/kg, days 0, 7, 14, 21, 28 and 35), cisplatin (4 mg/kg, days 0 and 7), LDN and taxol, LDN and cisplatin, or an equivalent volume of saline (daily). These dosages were selected based on published reports. 8,10,21,22,36,37 To ensure that all mice were injected an equivalent number of times, animals not assigned to receive treatment on a given day were administered saline. In groups receiving combined therapy, LDN was administered first. Taxol was dissolved in DMSO (10−2 mol/L) and further diluted in saline, while LDN and cisplatin were dissolved in saline; all drugs were prepared weekly.
Termination day measurements
According to the IACUC guidelines, the study was terminated when tumors became ulcerated or grew to 2 cm in diameter. All mice were euthanized by an overdose of sodium pentobarbital (100 mg/kg) and cervical dislocation 35 d following initiation of treatments. For examination of DNA synthesis in tumors, a subset of mice from each group was injected intraperitonealy twice with 100 mg/kg BrdU (bromodeoxyuridine) (Sigma-Aldrich, St Louis, MO, USA) at six and three hours prior to euthanasia. Tumors and spleens were removed and weighed, and the lymph nodes, liver and spleen examined for metastases. Tumor tissue was assessed for expression of OGF and OGFr, cell survival, angiogenesis and DNA synthesis.
Semiquantitative immunohistochemistry
Immunohistochemistry was utilized to evaluate the presence and relative levels of OGF and OGFr in tumor tissue following published procedures. 5,38,39 Tumors were excised, frozen in chilled isopentane, sectioned at 10 μm, fixed, permeabilized, and stained with antibodies to OGF and OGFr that were generated and characterized in our laboratory. 40 Images were taken at the same exposure time with care not to photobleach samples. A random sample of at least 10 fields/section, two sections/tumor and three tumors/group were evaluated. Controls were incubated with secondary antibodies only.
Protein isolation and Western blotting
Expression of OGFr was evaluated in tumors by Western blotting following published procedures. 5 Briefly, tissue was homogenized in RIPA buffer containing a cocktail of protease and phosphatase inhibitors (Roche, Indianapolis, IN, USA). Protein (60 μg) was subjected to 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred onto nitrocellulose and probed with antibodies to OGFr (1:200). Optical densities were normalized to β-actin (1:5000, Sigma-Aldrich), and the percent change in expression was calculated by dividing the normalized values of experimental samples to that of saline controls. Means and SE were determined from two independent experiments.
OGFr binding assays
Tumors were assayed for OGFr binding using custom synthesized [3H]-[Met5]-enkephalin (Perkin Elmer, Waltham, MA, USA; 52.7 Ci/mmol) following published procedures. 5,38,39 Saturation binding isotherms were generated using GraphPad Prism software (La Jolla, CA, USA), and independent assays were performed at least three times.
Mechanism of growth inhibition: DNA synthesis, angiogenesis, apoptosis and necrosis
Cells were assayed for DNA synthesis, necrosis and apoptosis, whereas tumor tissue was evaluated for DNA synthesis, apoptosis and angiogenesis. To measure DNA synthesis, cells were treated with 30 μmol/L BrdU for three hours prior to fixation, while tumors from mice receiving BrdU on the day of sacrifice were fixed in formalin overnight, processed in paraffin and sectioned at 10 μm. Preparations were processed with antibodies to BrdU (1:200; Invitrogen, Carlsbad, CA, USA) 5,38,39 to assess DNA synthesis, stained with hematoxylin/eosin 41–44 to evaluate endothelial cell-lined vessels containing red blood cells, or processed for TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) according to the manufacturer's instruction to measure apoptosis (Trevigen, Gaithersburg, MD, USA). For cells in tissue culture, the proportion of BrdU- or TUNEL-positive cells was determined for at least 500 cells on two coverslips/treatment group. For tumors, the proportion of BrdU-positive cells, number of TUNEL-positive cells and blood vessel density were determined from at least 10 random fields around the periphery of each tumor, with at least two sections/tumor and two tumors/treatment group evaluated. BrdU- and TUNEL-positive cells were counted in a 0.003-mm2 area, while blood vessel density was determined in a 0.16-mm2 area.
Chemicals
NTX was obtained from Sigma-Aldrich. Cisplatin was purchased from Alexis Biochemicals (Lausen, Switzerland), and taxol was obtained from Toronto Research Chemicals (North York, ON, Canada).
Statistical analysis
All data were analyzed using one-way analysis of variance, with subsequent comparisons made using Newman–Keuls tests (Graph Pad Prism Software). In some cases, data were evaluated using unpaired t-tests. P values <0.05 were considered to be significant.
Results
Combination of short-term NTX treatment with taxol or cisplatin provides an additive inhibitory effect on ovarian cancer cell number: in vitro studies
To establish the effects of short-term treatment of NTX on the growth of human ovarian cancer cells, SKOV-3 cultures were treated with 10−5 mol/L NTX for six hours every other day; this dosage and regimen inhibited the proliferation of SKOV-3 cells by 28–42% between 48 and 120 h. However, in cultures treated with continuous NTX, the cell number was increased 19–31% between 48 and 120 h, relative to sterile water treated cells, and increased 69–109% between 48 and 120 h, relative to cultures treated with short-term NTX (Figures 1a and b).
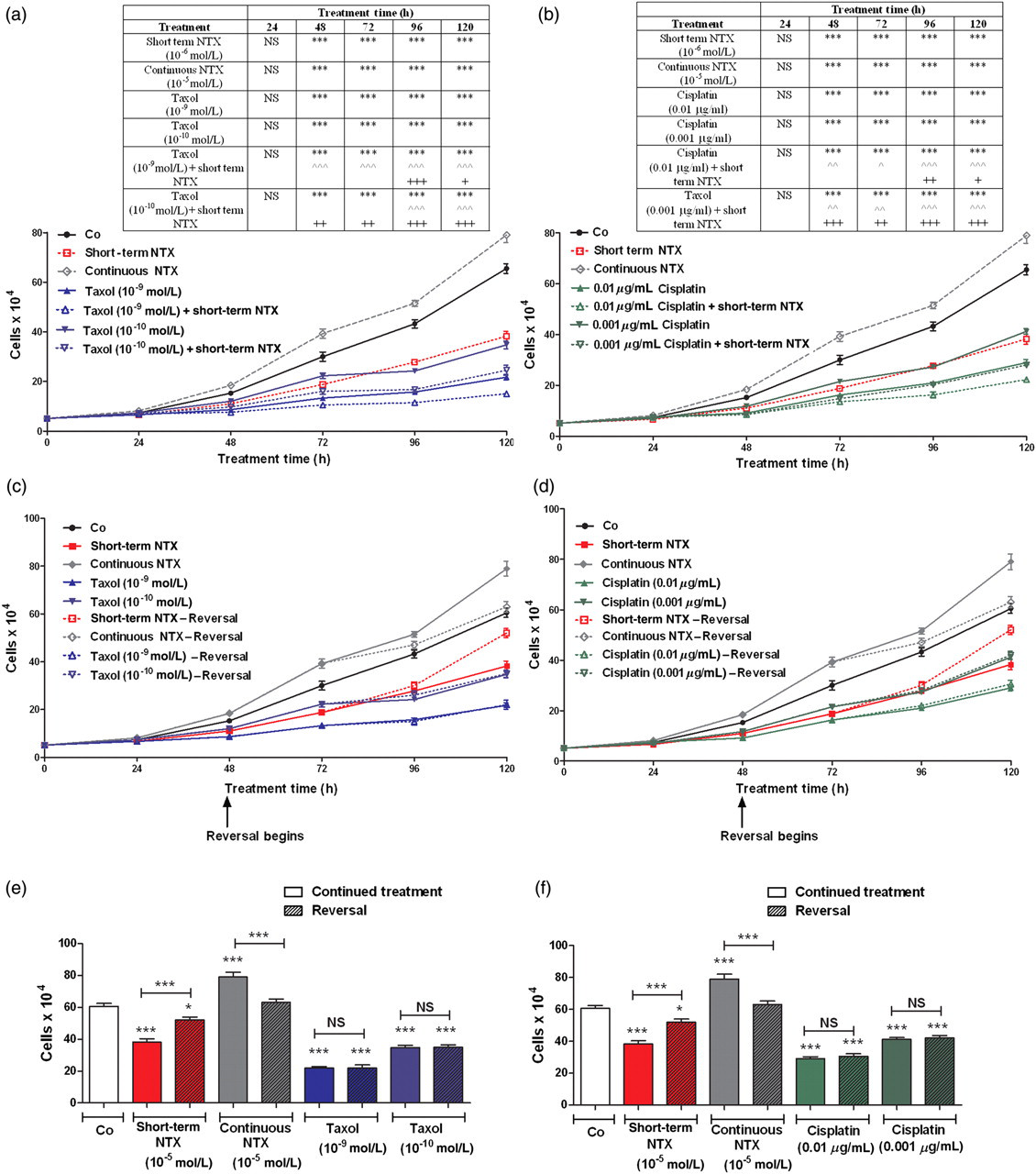
The combination of short-term naltrexone (NTX) treatment with taxol or cisplatin provides an additive inhibitory effect on ovarian cancer cell number in vitro. (a, b) Growth curves of SKOV-3 cells subjected to (a) short-term NTX (10−5 mol/L, 6 h every other day) and/or taxol (10−9 or 10−10 mol/L), continuous NTX (10−5 mol/L), or an equivalent volume of sterile water, or (b) short-term NTX and/or cisplatin (0.01 or 0.001 μg/mL), continuous NTX or an equivalent volume of sterile water over a 120-h period. Media and compounds were replaced daily unless otherwise noted. (c, d) Growth curves of SKOV-3 cells in reversibility experiments treated with short-term NTX, continuous NTX, taxol (10−9 or 10−10 mol/L) or cisplatin (0.01 or 0.001 μg/mL). At 48 h, a subset of cultures had media replaced without drugs (Reversal). Compounds and media were replaced daily unless otherwise indicated. (e, f) Cell number at 120 h in cultures from the reversibility experiments. Data represent cell counts (means ± SE) for at least two aliquots/well and two wells/treatment/timepoint. Two independent experiments were performed. Significantly different from sterile water-treated groups at *P < 0.05 and ***P < 0.001, from short-term NTX treatment alone at ∧ P < 0.05, ∧∧ P < 0.01 and ∧∧∧ P < 0.001, and from taxol or cisplatin at + P < 0.05, ++ P < 0.01 and +++ P < 0.001. NS = not significant (A color version of this figure is available in the online journal)
To ascertain the repercussions of combining a short-term NTX treatment with taxol, cells were exposed to these agents alone and in combination. The dosages of taxol (10−9 and 10−10 mol/L) were selected based on preliminary experiments that revealed no logarithmic growth at higher concentrations. Taxol at either concentration inhibited the number of SKOV-3 cells by 21–67% (Figure 1a). In cultures receiving short-term NTX (every 48 h) with taxol (at either concentration), cell number was reduced 36–61% compared with cells exposed to short-term NTX alone, and reduced 19–31% relative to cells treated with either concentration of taxol alone. At all time points evaluated over a 120-h period, an equivalent number of cells was noted in cultures receiving a combination of short-term NTX and taxol (10−10 mol/L) compared with those receiving the higher concentration of taxol (10−9 mol/L) alone.
To investigate the consequence of coupling a short-term NTX treatment with cisplatin, log phase cultures were subjected to these agents alone and in combination. The dosages of cisplatin (0.01 and 0.001 μg/mL) were chosen in view of preliminary experiments that revealed no logarithmic growth at higher concentrations. Cisplatin at either concentration suppressed the number of ovarian cancer cells by 23–51% (Figure 1b). In cultures receiving a short exposure to NTX every 48 h in combination with cisplatin at either concentration, cell number was reduced 21–42% compared with cells treated with this same regimen of NTX alone, and 23–32% relative to cells subjected to either concentration of cisplatin alone. At all time points evaluated, an equivalent number of cells was detected in cultures receiving short-term NTX and cisplatin (0.001 μg/mL) compared with those receiving the higher concentration of cisplatin (0.01 μg/mL) alone.
The growth effects of short-term exposure to NTX, but not to continuous treatment with taxol or cisplatin, are reversible
To study whether the effects of NTX given for a short or continuous duration, as well as either taxol or cisplatin, on growth could be reversed by withdrawing cells from drug exposure, SKOV-3 cultures were treated with short-term NTX (10−5 mol/L) every 48 h, continuous NTX (10−5 mol/L), taxol (10−9 or 10−10 mol/L), cisplatin (0.01 or 0.001 μg/mL), or an equivalent volume of sterile water. At 48 h, media and compounds were removed from all cultures and replaced with either media containing compounds or media lacking these agents (i.e. reversal). At 96 and 120 h, the short-term NTX-reversal group had 8% and 36% more cells, respectively, than in the group continuing to receive six hours of exposure to NTX every 48 h (Figures 1c–f). However, cultures in the continuous NTX-reversal group had 9% and 20% less cells at 96 and 120 h, respectively, than cells maintained on NTX. The taxol and cisplatin reversal groups did not differ from cultures continuing to be treated with taxol or cisplatin, respectively.
Mechanism of enhanced growth inhibition by the combination of short-term NTX with taxol or cisplatin: in vitro studies
Examination of apoptosis and necrosis in SKOV-3 cells treated with NTX (10−5 mol/L) for either a short- or a long-term duration, taxol (10−9 or 10−10 mol/L), cisplatin (0.01 or 0.001 μg/mL), or a combination of short-term NTX with taxol or cisplatin (at either concentration of these agents), revealed less than 0.1% positive cells for apoptosis or necrosis; these data were comparable to that obtained with cells subjected to sterile water (data not shown).
In regard to DNA synthesis, cells treated with NTX for a short term had 38% fewer cells labeled with BrdU than sterile water controls (Figures 2a and b). In contrast, cells subjected to NTX continuously had a 42% increase in BrdU labeling from cultures subjected to water. The combination of short-term exposure to NTX with taxol (at either concentration) reduced BrdU labeling by 47–49% from the group receiving taxol alone (Figure 2a). The coupling of NTX given for a short duration with taxol at either concentration suppressed DNA synthesis 39–47% from cells receiving NTX alone. Cultures receiving a short-term exposure to NTX in combination with cisplatin (at either concentration) had 38–44% decreased BrdU labeling indexes in contrast to cultures receiving only NTX, and 27–52% reduced labeling indexes relative to cultures receiving cisplatin alone (Figure 2b).

Mechanism of enhanced growth inhibition by the combination of short-term naltrexone (NTX) treatment with taxol or cisplatin. (a, b) Evaluation of DNA synthesis by bromodeoxyuridine (Brdu) labeling in SKOV-3 cells treated with short-term NTX (10−5 mol/L, 6 h every other day), continuous NTX (10−5 mol/L), taxol (10−9 or 10−10 mol/L), cisplatin (0.01 or 0.001 μg/mL), short-term NTX in combination with taxol or cisplatin, or an equivalent volume of sterile water (Co) for 120 h. Media and compounds were replaced daily unless otherwise indicated. Cells were pulsed with BrdU (30 μmol/L) for three hours prior to fixation and processed for BrdU immunoreactivity. Values represent the percent of cells labeled with BrdU (means ± SE) from at least 500 cells on 10 fields/coverslip and two coverslips/treatment group. Significantly different from Co at ***P < 0.001, from short-term NTX treated cells at ∧∧ P < 0.01 and ∧∧∧ P < 0.001, and from taxol or cisplatin alone at ++ P < 0.01 and +++ P < 0.001 (A color version of this figure is available in the online journal)
LDN inhibits established ovarian cancer, and has an additive inhibitory effect on tumor progression in combination with cisplatin
Beginning two days after initiation of treatments and persisting throughout the study, tumor volumes in mice with established subcutaneous ovarian xenografts were reduced by treatment with LDN (21–48%), taxol (21–54%), cisplatin (24–54%), LDN and taxol (39–60%), or LDN and cisplatin (16–60%) compared with control animals receiving saline (Figures 3a and b). The reductions in tumor volumes in mice subjected to LDN, taxol, cisplatin, or the combination of LDN and taxol were comparable. However, animals administered both LDN and cisplatin had tumor volumes that were reduced 9–30% from mice treated with LDN alone beginning on day 14, and were decreased 15–37% from mice receiving cisplatin alone beginning on day 16 (Figure 3b).

Growth of subcutaneous xenografts with SKOV-3 cells in mice treated with low dosage of naltrexone (LDN), taxol, cisplatin, or LDN in combination with taxol or cisplatin chemotherapy. When tumors became visible (day 0), animals were injected with either LDN (0.1 mg/kg, daily), taxol (3 mg/kg, days 0, 7, 14, 21, 28 and 35), cisplatin (4 mg/kg, days 0 and 7), LDN and taxol, LDN and cisplatin, or an equivalent volume of saline (daily). (a, b) Tumor volumes were assessed three times/week. Data for the saline and LDN groups in (a) and (b) are from the same groups of mice. (c) Representative images of tumors before and after removal from animals following 35 d of treatment. (d) Terminal tumor volume (mm3). (e) Terminal tumor weight (g). Values represent means ± SE for 10 mice/group. Significantly different from saline at *P < 0.05, **P < 0.01 and ***P < 0.001, from LDN at *P < 0.05 and **P < 0.01, and from cisplatin at + P < 0.05, ++ P < 0.01 and +++ P < 0.001 (A color version of this figure is available in the online journal)
On the day of termination (day 35), mice from all treatment groups displayed a visible reduction in tumor size (Figure 3c) compared with controls subjected to saline, with decreases in both tumor volume (43–55%; Figure 3d) and tumor weight (28–46%; Figure 3e) recorded. The reductions in tumor volume and weight in mice exposed to the combination of LDN and taxol were comparable to those in animals subjected to LDN or taxol alone (Figures 3d and e). However, relative to mice treated with cisplatin alone, the combination of LDN and cisplatin depressed tumor volumes and tumor weights by 26% and 25%, respectively. Mice receiving the combination of LDN and cisplatin had terminal tumor volume and weight that were equivalent to mice treated with LDN alone.
Effects of treatments on body weight and gross observations
Although all mice weighed approximately 18–20 g at the beginning of the experiment (Figure 4), mice receiving cisplatin on days 0 and 7 had a 7–12% reduction in body weight compared with saline administered controls on days 2, 9 and 11 of the study. Mice receiving the combination of LDN and cisplatin also were reduced from saline-treated controls in body weight (8%) on day 2 of the study. However, cohorts exposed to both LDN and cisplatin were comparable in body weight to saline-administered controls on days 9 and 11 of the study. Body weights of animals administered LDN, taxol or the combination of LDN and taxol were similar throughout the study to saline controls (Figure 4).
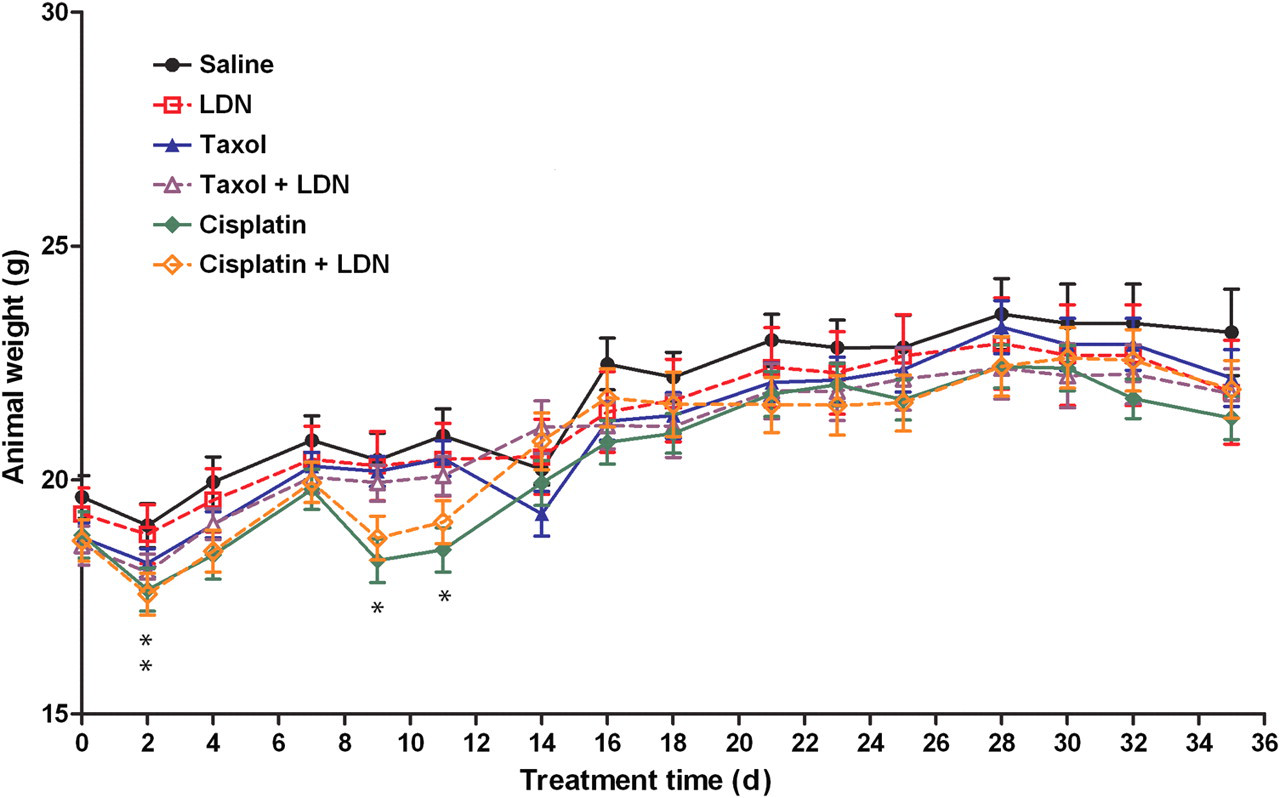
Body weights of mice with subcutaneous xenografts of ovarian cancer cells treated with low dosage of naltrexone (LDN), taxol, cisplatin, or LDN in combination with taxol or cisplatin; treatments were initiated when tumors were visible. Values represent means ± SE body weight for 10 mice/group. The cisplatin group was significantly different from mice subjected to saline at *P < 0.05 on days 2, 9 and 11, and animals receiving both LDN and cisplatin were significantly different from saline at *P < 0.05 on day 2 (A color version of this figure is available in the online journal)
Spleen weights on the day of termination did not differ between any group of mice (data not shown), and behavioral abnormalities were not evident. Metastases or lesions were not detected in mice from any group.
Mechanism of enhanced growth inhibition by LDN with cisplatin: effects on apoptosis, DNA synthesis and angiogenesis
Examination of apoptosis by the TUNEL assay revealed similar levels of programmed cell death in tumors taken from mice treated with LDN or saline (Figure 5a). However, animals treated with taxol or cisplatin, either alone or in combination with LDN, had approximately a three-fold greater number of apoptotic cells in tumors than that of saline-administered controls.

Mechanism of tumor growth inhibition by treatment with low dosage of naltrexone (LDN), taxol and/or cisplatin: effects on apoptosis, DNA synthesis and angiogenesis. Treatments were initiated when tumors were visible (day 0) and tumor tissue was assessed 35 days later. (a) Number of apoptotic cells per 0.003 mm2, as measured by the TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay. (b) Percentage of cells with bromodeoxyuridine (Brdu) labeling. (c) Number of blood vessels per 0.16 mm2, as assessed by hematoxylin/eosin staining to identify endothelial cell-lined blood vessels. Values represent means ± SE determined from at least 10 random fields from the periphery of two tumor sections/mouse and two mice/group. Significantly different from saline at ***P < 0.001, from LDN at ∧ P < 0.05 or ∧∧∧ P < 0.001, and from cisplatin by + P < 0.05 or ++ P < 0.01 (A color version of this figure is available in the online journal)
With respect to cells in tumors undergoing DNA synthesis, a reduction of 42–57% was noted in all treatment groups compared with mice injected with saline (Figure 5b). The BrdU labeling index in tumors from animals receiving the combination of LDN and taxol was comparable to cohorts subjected to either LDN or taxol alone, while the BrdU labeling index in tumors from animals receiving both LDN and cisplatin was reduced 24% compared with subjects treated with cisplatin alone. Levels of DNA synthesis in tumors were similar in mice treated with only LDN or the combination of LDN and cisplatin.
With respect to the density of blood vessels in tumors, blood vessel density was reduced 52–73% in all treatment groups relative to animals exposed to saline (Figure 5c). Blood vessel density in tumors from mice receiving the combination of LDN and taxol was comparable to cohorts treated with LDN or taxol alone. In contrast, blood vessel density was decreased 42–44% in subjects treated with the combination of LDN and cisplatin compared with animals administered LDN or cisplatin alone.
The presence and expression of OGF and OGFr in ovarian cancer xenografts
To evaluate OGF distribution and relative expression levels in xenografts of ovarian tumors, semiquantitative immunohistochemistry was performed. OGF was visible in the cytoplasm and a speckling of immunoreactivity was often noted in cell nuclei (Figure 6a). Tumors processed with only secondary antibody showed no staining (Figure 6a, inset). OGF distribution did not differ between groups. However, OGF immunofluorescence (mean gray value) in mice treated with LDN, both LDN and taxol, or the combination of LDN with cisplatin was increased 33–39% relative to animals administered saline (Figure 6b). Comparable levels of OGF immunofluorescence were noted in tumors from mice treated with saline, taxol or cisplatin.

The distribution and expression of opioid growth factor (OGF) and opioid growth factor receptor (OGFr) in xenografts of SKOV-3 cells. Mice were treated with low dosage of naltrexone (LDN), taxol, cisplatin or LDN in combination with taxol or cisplatin beginning when tumors were visible. (a, c) Photomicrographs taken at the same exposure time of tumors on the day of sacrifice (day 35). Sections were stained with antibodies (1:200) to OGF (a) or OGFr (c). Rhodamine conjugated IgG (1:1000) served as the secondary antibody and nuclei were visualized with 4′,6-diamidino-2-phenylindole. Preparations incubated with secondary antibodies only (insets). Bar = 10 μm. (b, d) Semiquantitative measurement of OGF (b) and OGFr (d) staining intensity (mean gray value) from at least 10 fields from two sections/tumor and three mice/group. (e, f) Western blot of the 62 kDa band of OGFr (e) and densitometric analysis (f) normalized to β-actin from two independent experiments. (g) Saturation isotherms calculating the binding capacity (B max) of OGFr in xenografts from at least three independent assays performed in duplicate. Data represent means ± SE. Significantly different from saline at *P < 0.05, **P < 0.01 and ***P < 0.001 (A color version of this figure is available in the online journal)
To examine OGFr distribution and relative expression, immunohistochemistry, Western blotting and receptor binding assays were performed on xenografts. The cellular location of OGFr was similar in all groups, with immunoreactivity detected in the cytoplasm and nucleus (Figure 6c). Tumors processed with only secondary antibody showed no staining (Figure 6c, inset). Relative to saline-administered controls, OGFr expression in mice treated with LDN, LDN and taxol, or LDN and cisplatin was increased 46–61% using semiquantitative immunohistochemistry (Figure 6d). Further evaluation of OGFr expression using Western blotting showed that mice treated with LDN had an 87% increase in OGFr expression in their tumors compared with saline-administered controls (Figures 6e and f).
Receptor binding assays indicated specific and saturable binding for OGFr in tumors of all groups, with a one site model of binding recorded (Figure 6g). Binding capacity (B max) values were markedly increased (112–136%) in mice treated with LDN, LDN and taxol, or LDN and cisplatin, compared with control animals receiving saline. However, binding affinity (K d) for OGFr did not differ among treatment groups and ranged from 2.7 to 6.2 nmol/L (data not shown).
Discussion
The present study demonstrates for the first time that exposure to NTX for a short duration suppresses cell proliferation and DNA synthesis in vitro. This is in distinct contrast to a long-term continuous exposure to NTX, which accelerates cell proliferation and DNA synthesis of human ovarian cancer cells, a result reported previously. 5,6 The effects of a short-term treatment with NTX on proliferation of ovarian cancer cells was comparable to that of the reductions recorded herein and elsewhere with taxol or cisplatin. 45,46 When NTX in a regimen of short-term exposure was combined with taxol or cisplatin under in vitro conditions, the effects on cell replication were greater than with the individual drugs. Indeed, the effects of a 10−9 mol/L or 0.01 μg/mL concentration of taxol or cisplatin, respectively, could be achieved with a 10-fold lower concentration of either agent when combined with a short-term treatment with NTX. The inhibitory influence on cell proliferation by short-term treatment with NTX was reversible, with cell number having a trajectory returning to control levels upon withdrawal of this opioid antagonist. In contrast, discontinuation of taxol or cisplatin treatment by providing fresh media without either drug did not change cell kinetics, with these cultures resembling those that continued to receive either drug. These results show that the combination of two treatment modalities, NTX given for a short duration and a chemotherapeutic agent, act in an additive fashion to impede the growth of human ovarian cancer cells in tissue culture.
The results of this study make the seminal observation that intermittent opioid receptor antagonism, achieved by a daily exposure to LDN, markedly impedes the progression of human ovarian tumorigenesis. Treatment with LDN resulted in reductions in both tumor volume and weight that were approximately one-third of those for tumor-bearing animals injected with saline. Moreover, the magnitude of effects of LDN on tumorigenicity was equal to that recorded with either of two standard of care agents: taxol and cisplatin. When LDN was combined with cisplatin, but not with taxol, even greater antitumor activity than either agent alone was recorded. This is in contrast to the results obtained in vitro, wherein a short-term exposure to NTX could be combined with either taxol or cisplatin for an enhanced inhibitory effect on growth. This is not the first time, however, that a discrepancy in the sensitivity to a chemotherapeutic agent has been observed when testing drugs under in vitro and in vivo conditions. 47 In summary, these results provide evidence that LDN has a potent antitumor effect on ovarian carcinogenesis, and reveal that a combination of biotherapeutic modulation with LDN and chemotherapy with cisplatin have a cooperative effect in retarding the growth of this lethal disease.
Although both LDN and/or taxol in the concentrations and regimens used in this study were not overtly toxic to mice with xenografts of ovarian cancer, animals subjected to cisplatin alone had notable reductions in body weight on several days in the study. This systemic toxicity from cisplatin was diminished by simultaneous administration with LDN, indicating that this regimen with an opioid antagonist has the capacity to protect against toxicological insults. The amelioration of cisplatin-induced toxicity by LDN, however, was not accompanied by a diminution of the antitumor action of cisplatin. In fact, the combination of LDN and cisplatin had an effect on tumor growth (i.e. weight, volume) that exceeded cisplatin alone. The mechanism of protection afforded by LDN against cisplatin toxicity is unknown. The alleviation of toxicity of one agent by the administration of another drug, however, is not without precedence. 48,49 The finding of protection afforded by LDN from the side-effects of cisplatin may allow higher doses of cisplatin to be administered to improve the therapeutic efficacy of this agent. This may be advantageous, as the success of chemotherapy is often limited by resistance of cancer cells as well as toxicity, and the possibility of increasing the concentration of drugs without an accompanying increase in adverse events could be extremely beneficial.
The mechanism for enhanced growth inhibition of SKOV-3 cells in tissue culture by a combination of short-term NTX with either taxol or cisplatin was related to DNA synthesis but was not associated with induction of apoptosis or necrosis, at least at the low dosages of taxol and cisplatin used herein. Under in vivo conditions, however, the combination of LDN and cisplatin, but not the coupling of LDN and taxol, had an additive effect on tumorigenic events. Both taxol and cisplatin induced apoptosis, depressed cell proliferation and reduced the number of blood vessels. LDN, on the other hand, altered cell proliferation and angiogenesis, but did not influence apoptosis. The mechanism of the enhanced effect of LDN and cisplatin on ovarian cancer appears to be related to the number of cells undergoing DNA synthesis, as well the density of blood vessels, when these agents are combined. The lack of enhanced activity of a combination of LDN and taxol on tumorigenesis appears to be correlated with a failure of these agents to have an additive action on cell proliferation or angiogenesis. The effects of these agents are consistent with previous observations. Taxol and cisplatin are well known to induce cell death in a cell-phase-specific manner at the G2/M phase through binding to and stabilizing microtubules, 50 and binding to DNA and nuclear proteins to form intra- and interstrand cross-links, 51,52 respectively. The demonstration that LDN inhibits DNA synthesis to reduce tumorigenesis is also consistent with a previous study where mice transplanted with murine neuroblastoma were treated with LDN. 10 In that study, DNA synthesis was initially increased during the period of opioid receptor blockade (4–6 h), but was markedly depressed in the subsequent 18–20 h interval when NTX was no longer present, resulting in a net inhibition of cell proliferation and tumorigenic events. 10 Thus, the individual effects of biotherapy and chemotherapy can be enhanced by combining agents that target similar and differing fundamental biological processes.
In all previous studies investigating the effects of an intermittent opioid receptor blockade with LDN on carcinogenesis, the paradigm used was to initiate administration of opioid antagonist concomitant with tumor cell inoculation. 8,10,21,22 These reports concluded that the effects of LDN were pronounced as to altering the early events of tumor development (latency to initiation) without affecting events subsequent to appearance (tumor growth). 2,8,10,21 The present study, however, reveals for the first time that LDN functions to inhibit ovarian cancer progression in mice with established tumors. These results reveal that LDN not only can influence early tumorigenic events but can also exert a potent action on an established cancer. With these observations in mind, it may be conjectured that LDN can be used as an antitumor agent in ovarian cancer prior to tumor expression and serve as a prophylactic therapy. Moreover, our present findings would suggest that patients with established disease or following tumor resection could benefit from LDN biotherapy alone or in combination with standard of care drugs.
A number of lines of evidence from previous reports, as well as the present investigation, suggest that the opioid peptide–opioid receptor system involved with the inhibitory action of both short-term exposure to NTX in vitro, and intermittent opioid receptor blockade with LDN in vivo, is the OGF–OGFr axis. First, among a panel of natural and synthetic opioid peptides, many specific for the classic μ, δ or κ opioid receptors, OGF was the singular opioid peptide with growth inhibitory properties on ovarian cancer cell proliferation. 5 Second, the effect of OGF on depressing the proliferation of ovarian cancer cells was eliminated by concomitant exposure to the short-acting opioid antagonist naloxone, demonstrating that OGF action was mediated by an opioid receptor. 5 Third, using siRNA technology, knockdown of OGFr stimulated cell proliferation and neutralized the repercussions of exogenous OGF exposure, suggesting that the effects of endogenous and exogenous OGF are dependent on this non-classical opioid receptor. 5,6 Fourth, administration of NTX is known to upregulate both OGF and OGFr. 20,28 Fifth, continuous exposure of ovarian cancer cells to NTX accelerated cell proliferative events, suggesting that the effect of OGF not only utilized an inhibitory pathway but was tonically active in maintaining the homeostatic balance of cell replicative events. 5,6 Sixth, short-term opioid antagonism (i.e. LDN) initially elevates DNA synthesis during the period of opioid receptor blockade, and depresses DNA synthesis when NTX is no longer present. 10 In view of previous evidence 5,6,10,20,28 showing that NTX targets cell proliferative pathways, the regimen of a short-term exposure to this opioid antagonist appears to have led to an enhanced interaction of the upregulated OGF and OGFr in the interval when NTX was no longer present. Thus, short-term NTX-mediated modulation of the OGF–OGFr axis appears to account for the depressed DNA synthesis and proliferation of human ovarian cancer cells under in vitro conditions. Moreover, LDN's antitumor effect in established xenografts of ovarian cancer was shown to upregulate the expression of OGF and OGFr. Therefore, we would conjecture that this upregulation of the OGF–OGFr axis by LDN intensifies the interaction of OGF and OGFr when NTX is no longer present. The net effect of LDN, therefore, is to have an exaggerated inhibitory influence on the progression of ovarian cancer. In view of these arguments, we postulate that the effects of a short-term exposure to NTX in tissue culture, and the influence of LDN on tumor progression in mice, have a common denominator in terms of mechanism: the OGF–OGFr axis.
With opioid receptor antagonism serving as a means to manipulate the opioid system related to growth, the OGF–OGFr axis, this study demonstrates that endogenous opioids are determinants of ovarian carcinogenesis and concur with previous reports. 5,6 These findings raise the exciting potential in the clinical setting of utilizing LDN biotherapy alone, or in combination with chemotherapy, as novel treatment modalities for ovarian cancer. LDN has been documented to be safe for administration in humans with Crohn's disease 12,53 or multiple sclerosis. 54–56 LDN has not been tested in cancer patients for safety or efficacy. However, these reports documenting the lack of toxicity of LDN in humans, and our preclinical findings that LDN is efficacious in suppressing ovarian cancer in vivo, makes the transition from the laboratory to the clinic in terms of using LDN alone or in combination with standard of care drugs feasible for the treatment of ovarian cancer. Should LDN prove to be an effective treatment for ovarian cancer, there are a number of advantages this agent would have over standard of care chemotherapies. LDN is orally effective, inexpensive and not associated with toxic side-effects. 12,53–56 Our results suggest that LDN could be used under three different circumstances: (i) as a prophylactic agent, particularly in patients with a family history of ovarian cancer, (ii) as a first-line treatment, alone or in combination with standard of care drugs, following cytoreductive surgery, and (iii) following relapse when all other treatments are palliative.
Footnotes
ACKNOWLEDGEMENTS
We are grateful for the support by the Paul K and Anna E Shockey Family, Bonnie and Ken Shockey, and the Zagon/Kostel families.