
Abstract
Embryonic stem cells (ESCs) are pluripotent, self-renewing cells that are isolated during the blastocyst stage of embryonic development. Whether these cells are derived from humans, mice or other organisms, all ESCs must employ mechanisms that prevent the propagation of mutations, generated as a consequence of DNA damage, to somatic cells produced by normal programmed differentiation. Thus, the prevention of mutations in ESCs is important not only for the health of the individual organism derived from these cells but also, in addition, for the continued survival and genetic viability of the species by preventing the accumulation of mutations in the germline. Induced pluripotent stem cells (IPSCs) are reprogrammed somatic cells that share several characteristics with ESCs, including a similar morphology in culture, the re-expression of pluripotency markers and the ability to differentiate into defined cell lineages. This review focuses on the mechanisms employed by murine ESCs, human ESCs and, where data are available, IPSCs to preserve genetic integrity.
Keywords
Introduction
Embryonic stem cells (ESCs) are derived from the blastocyst inner cell mass and are required for formation of the embryo proper. If cultured under optimal conditions, these cells can be grown indefinitely and remain pluripotent, a trait that is attributable to their symmetric cellular division. 1 ESCs were first derived and grown in culture from murine embryos nearly 30 years ago. 2,3 The initial work on mouse ESCs (mESCs) has led to the development of techniques that have made possible significant advancements for the scientific community, mainly through the generation of gene knockout and knockin technology. It was not until 17 years later that human ESCs (hESCs) were successfully obtained, 4 owing to differences in growth requirements, ethical controversies and overall complexities compared with mESCs. The most desirable attribute of hESCs is their ability to differentiate into any cell type, a feature that is currently being exploited in several phase I/II clinical trials focusing on regenerative medicine and tissue replacement therapies.
Induced pluripotent stem cells (IPSCs) were independently generated by several groups in 2006–2007, first from mouse cells 5–7 and later from human cells. 8,9 Through the introduction of combinations of several genes typically expressed during early embryogenesis, somatic cells could be ‘reprogrammed’ to mimic ESCs. These reprogrammed cells have laid the foundation for numerous applications including patient-specific tissue regeneration and drug tailoring, as well as disease modeling, without the ethical objections raised by the destruction of human embryos required to procure hESCs.
A hypothesis in the stem cell biology field is that ESCs (and possibly IPSCs) must employ mechanisms to ensure that genetic integrity remains pristine. The rationale for this proposition comes from the idea that the accumulation of mutations in ESCs would be detrimental to the developing organism and would negatively impact the genetic viability of the species from which the ESCs are derived, if mutations arise in those ESCs from which germ cells are derived. Several lines of evidence give credence to this postulate including: (1) that murine ESCs display lower mutation frequencies compared with somatic cell types (i.e. errors in replication/point mutation, etc.); (2) that ESCs with damaged DNA are eliminated from the self-renewing pluripotent population through apoptotic or differentiation programs; and (3) that ESCs demonstrate an elevated DNA repair capacity compared with somatic cells and preferentially utilize pathways that repair DNA damage with high fidelity. The remainder of this review will discuss these points as they relate to ESCs, IPSCs and somatic cells.
Mutational burden
ESCs must preserve genetic integrity to prevent the accumulation of mutations throughout the genome of the individual and to prevent the transmission of those mutations to subsequent generations. A study by Cervantes et al. 10 using mESCs demonstrated that this is indeed the case. Mouse ESCs display about a 100-fold lower spontaneous mutation frequency at the Aprt reporter locus (10−6) compared with isogenic mouse embryo fibroblasts (mEFs; 10−4) using a selection-based assay. When the mechanisms leading to this observation were examined, the majority of events were due to heterozygosity (LOH), with point mutations and deletions making up the remainder. In mEFs, the observed LOH was mainly due to mitotic recombination, whereas in mESCs, uniparental disomy predominated, followed by mitotic recombination. When mutation frequencies were measured at the Hprt locus in the same study as well as others, significantly fewer mutations were observed in the mESCs. 10–13 Since Hprt is located on the X chromosome, and the cells used in this study were derived from male embryos, there was no contribution of LOH to the observed spontaneous mutation frequencies. In addition to displaying a lower spontaneous mutation frequency, mESCs also incur mutations at a slower rate, 400-fold more slowly than mEFs. 10 Not all reports, however, support the observation that mESCs display lower mutation frequencies. For example, two studies have reported that mutation frequencies at the Rosa26 locus were similar between mESCs and mEFs, at a level of 10−4, using fluorescent protein reporter-based technologies. 14,15 Whether these findings are unique to the Rosa26 locus, or can be attributed to differences in the methods used to quantitate mutation frequencies, or perhaps to differences in the ESC lines remains unknown. Mutation frequencies in hESCs or IPSCs have not yet been explored, but one may assume that similarities do exist between all pluripotent cell types.
Cell cycle, DNA damage signaling and checkpoint control
Extensive research has demonstrated that the cell cycle profiles of mESCs are unique compared with somatic cell types. 16,17 For example, the proportion of mESCs occupying the S-phase of the cell cycle ranges from 50% to 70% in an asynchronous population, whereas in early passage mEFs, only about 20–30% of asynchronous cells are in this phase (Figure 1). The time required for mESCs to complete a full cell cycle is also short, ranging from 8 to 12 h, whereas non-transformed early passage primary murine cells have much longer cycling times of 24–36 h. 18
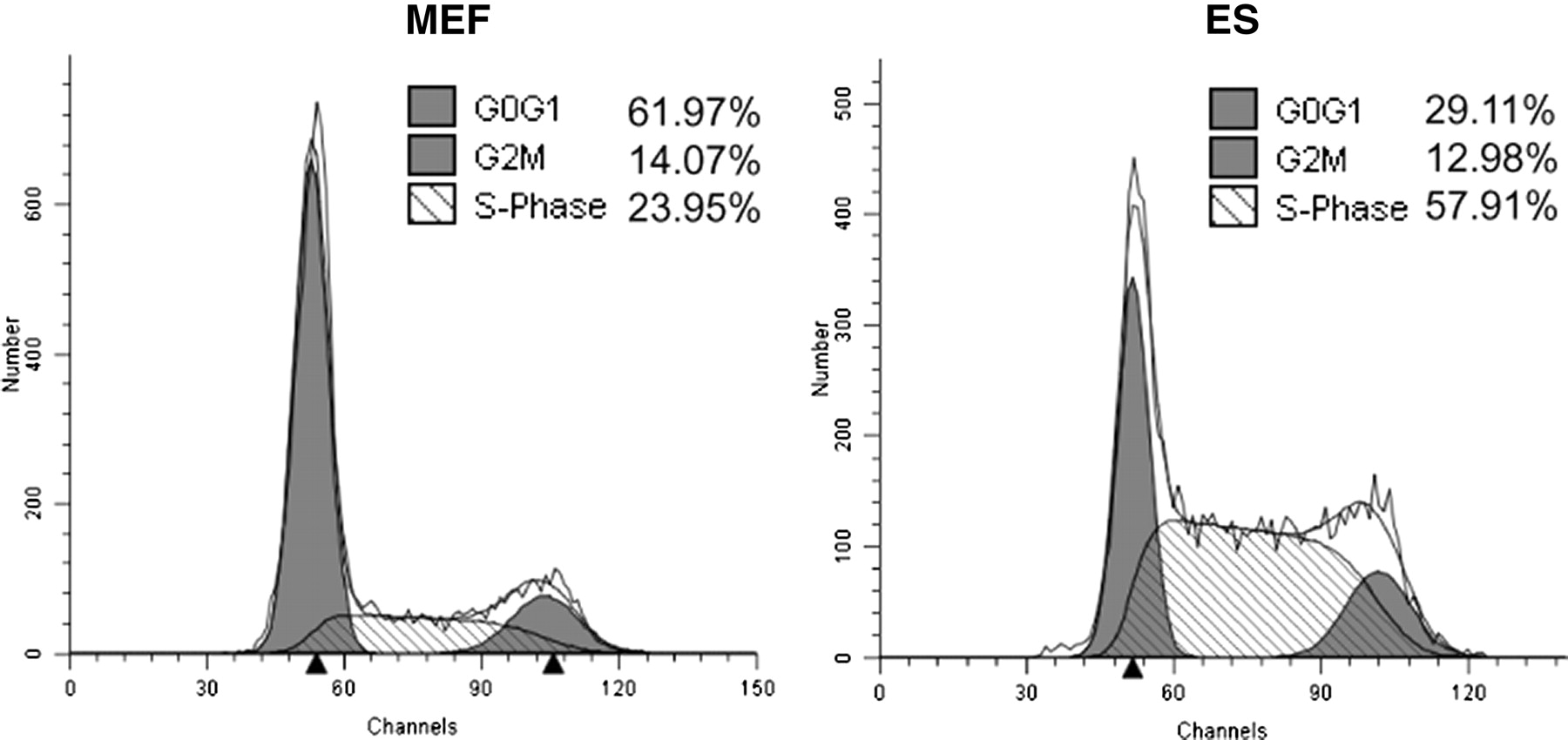
Cell cycle profiles of mouse embryo fibroblasts (MEF) and murine embryonic stem cells (ES). Asynchronous mESCs or early passage mEFs were grown to 60% confluency before harvest and fixation. Cells were treated with RNase A and stained with propidium iodide prior to analysis by flow cytometry for DNA content
The cell cycle distribution of ESCs derived from human embryos is similar to that of mESCs. Both display abbreviated gap phases and a large proportion of cells can be found in the S-phase. 17 However, unlike mESCs, hESCs have a total cycling time of 15–36 h, depending on culture conditions and passage number. 19,20 Human IPSCs derived from IMR-90 fibroblasts have a cell cycle profile similar to that of mESCs and hESCs, since about 70% of the cells are found in the S-phase at any given time, 21 and they have cell cycle times of about 16–18 h. 22
The responses of ESCs to DNA damaging agents are very different from those of somatic cell types. For example, mESCs lack a G1 checkpoint following DNA damage, whereas most somatic cells arrest in the G1 phase of the cell cycle after such damage is incurred. The lack of a G1 checkpoint in mESCs can be explained by two different mechanisms. One explanation is that in some studies, but not all, the p53 protein does not respond to DNA damage in a typical manner. Mislocalization of the protein in the cytoplasm prior to and after DNA damage prevents the transcription of p53 target genes, including the cell cycle inhibitor p21. 23,24 The other major pathway in the activation of the G1 checkpoint involves the checkpoint kinase protein Chk2. In addition to phosphorylating and stabilizing p53, Chk2 can phosphorylate the Cdc25a phosphatase and promote its degradation after DNA damage. With diminished Cdc25a protein, inhibitory phosphate groups on the cell cycle-dependent kinase Cdk2 are ineffectively removed, thus preventing entry of cells into the S-phase. In mESCs, the Chk2 protein is mislocalized to centrosomes, even under conditions of DNA damage, rendering it incapable of phosphorylating Cdc25a. 24 The resultant stabilization of Cdc25a purportedly promotes increased Cdk2 activity and thus mESCs do not arrest in G1. This study demonstrated that a G1 arrest could be successfully restored in these cells after exogenous expression of Chk2 and treatment with 10 Gy X-rays, which subsequently resulted in the degradation of Cdc25a. As expected, the lack of Cdc25a protein resulted in the prevention of Cdk2 activation.
In contrast with Hong et al., 24 another group reported that Cdc25a can be degraded after ionizing radiation (IR) treatment in mESCs, and that this degradation was not dependent on Chk2, but rather dependent on glycogen-synthase kinase 3β (Gsk3β). 25 Chemical inhibition of Gsk3β was sufficient to stabilize Cdc25a after IR treatment in mESCs, while little impact was seen in NIH 3T3 cells under the same conditions. Even though Cdc25a was degraded after IR treatment, the authors showed that this degradation had little impact on Cdk2 activity. However, inhibition of Cdk2 activity with olomoucine II after IR treatment in mESCs resulted in the accumulation of more cells in the G1 phase of the cell cycle, suggesting that the activity of Cdk2 was the driving force promoting the S-phase entry.
Contrary to what was reported in mESCs but similar to reports for somatic cells, CHEK2 in hESCs does not localize to centrosomes but remains exclusively nuclear. 26 Nuclear CHEK2 in these cells can phosphorylate CDC25A and target it for degradation after certain types of DNA damage such as that caused by ultraviolet (UV) radiation, resulting in a G1 arrest. In response to UV, CHEK2 can also phosphorylate and stabilize the p53 protein, which in turn increases the transcription of p21 mRNA. 26 However, hESCs do not display a G1/S checkpoint in response to all forms of DNA damage, such as that induced by IR. 27,28 Similar to hESCs, IPSCs also appear to lack an IR-induced G1 checkpoint and instead arrest in the G2 phase of the cell cycle. 21
Hypersensitivity to DNA damage, and cell death
Another mechanism ESCs may utilize to ensure a minimal mutational burden is to eliminate individual self-renewing cells with damaged DNA from the stem cell population. Support for this mechanism derives from independent observations which show that these cells are hypersensitive to DNA damaging agents. For example, treatment of mESCs with UV irradiation or methylating agents results in the massive induction of cell death. 29–31 We have observed a similar phenomenon in mESCs after treatment with the topoisomerase II inhibitor etoposide, where cell death occurs in mESCs at a dose 10 times lower than that needed to kill 3T3 cells (Figure 2). Human ESCs and IPSCs also appear hypersensitive to DNA damaging agents such as IR and they rapidly undergo caspase 3-dependent apoptosis after exposure. 21,27,28
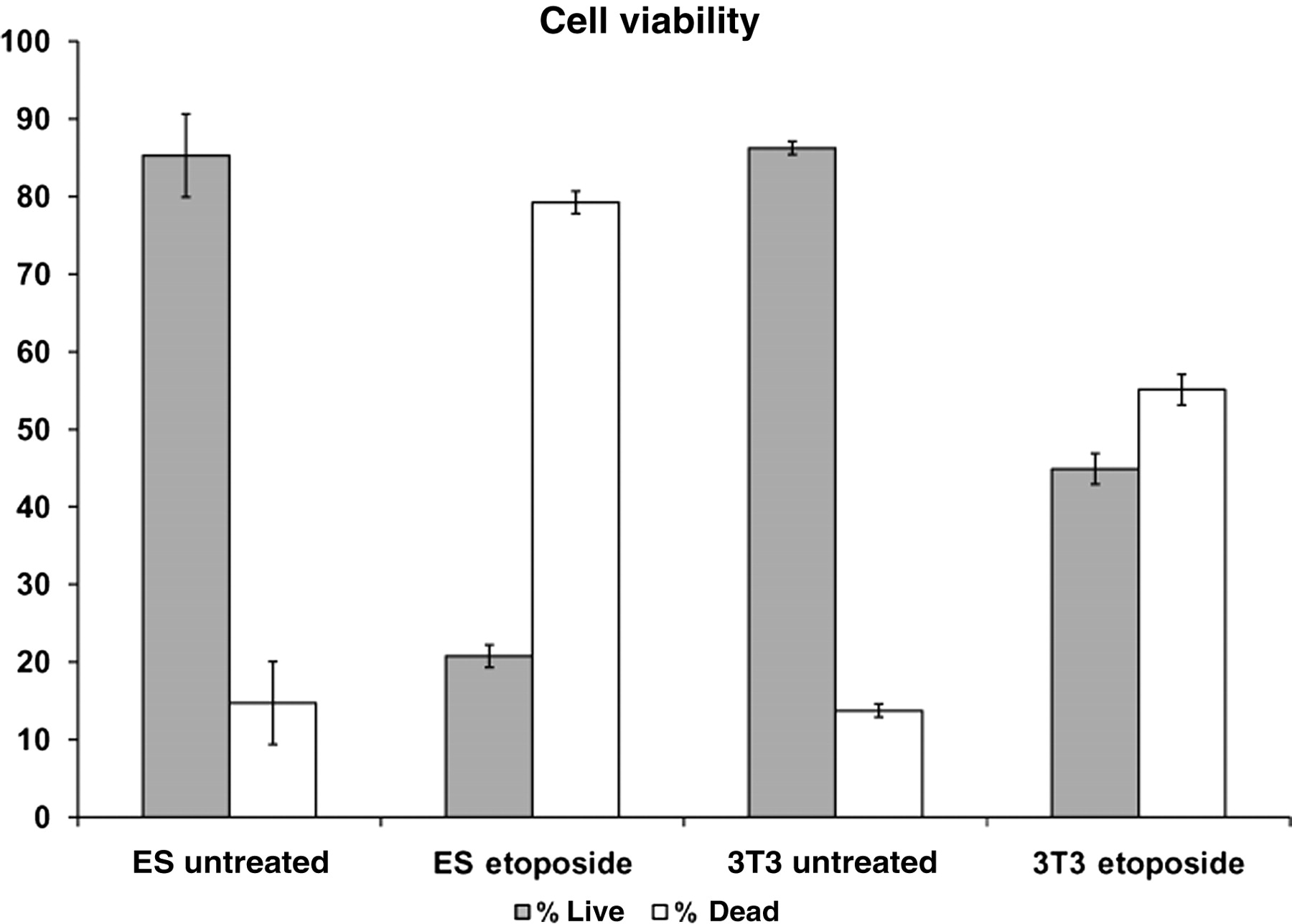
Murine embryonic stem cells (ES) are more sensitive to etoposide than Swiss 3T3 cells. ES or Swiss 3T3 cells were treated with either 10 or 100 μmol/L etoposide for 4 or 24 h, respectively. Annexin-V/PI staining was completed and data were obtained following flow cytometry analysis. Error bars represent the SEM
P53 protein: a trigger for differentiation?
Typically, the p53 protein is tightly regulated and kept at low levels in somatic cells. When cells experience oncogenic stress or DNA damage, the p53 protein is stabilized and functions to induce transient or permanent cell cycle arrest (senescence) or apoptosis through its transcriptional activities as well as its function localizing to and permeabilizing mitochondria. 32 An emerging idea in ESC biology is that a p53-dependent pathway may control differentiation in these cells under conditions of stress, providing an alternative mechanism by which to eliminate damaged cells from the pluripotent stem cell pool. As stated previously, some reports indicate that mESCs express stable p53 protein at high levels under basal conditions. These reports also suggest that the p53 protein localizes in the cytoplasm, 24,33 even under conditions of DNA damage. 24 Recently, however, evidence has been accumulating that suggests p53 can localize to the nucleus after DNA damage 33 and may activate signaling networks linked to differentiation. When wild-type mESCs are treated with UV or doxorubicin, for example, the p53 protein is reported to translocate to the nucleus and bind the Nanog promoter, causing downregulation of Nanog mRNA and subsequently reducing protein expression. 34 Nanog protein expression is a critical factor in maintaining pluripotency and self-renewal in ESCs, and lowered protein expression induces differentiation programs. This differentiation phenomenon was not observed in p53 null mESCs after induction of DNA damage. In addition, as mESCs differentiate, p53 is post-translationally modified by phosphorylation to higher levels than found in undifferentiated cells. Notably, many of these sites are also phosphorylated after DNA damage in somatic cells. 35 This suggests that p53 modifications, which are important in DNA damage signaling and apoptosis in somatic cells, may play other roles in ESCs, such as the invocation of differentiation.
As with mESCs, the function and behavior of p53 in hESCs is also controversial. While some reports argue that p53 in hESCs functions like p53 in somatic cells following DNA damage, others suggest that p53 does not activate the typical transcriptional networks, such as p21, in response to DNA damage induction. In one study using hESCs, p53 protein was upregulated after treatment with 20 J/m2 UV, and the protein localized to both the nucleus and the mitochondria. 36 However, these cells induce an apoptotic program that is dependent on the localization of p53 to the mitochondria and do not demonstrate appreciable transcriptional activity of typical p53 target genes following DNA damage, unless the cells are induced to differentiate. Yet, knockdown of p53 expression with siRNA in hESCs has been shown to reduce the rate of spontaneous differentiation in these cells, adding credence to the authors' previous observations. 36 This study also demonstrated that like in mESCs, p53 binds to the Nanog promoter after DNA damage and downregulates the level of Nanog transcript. In contrast to the mESCs, the Oct4 protein in hESCs was also diminished through inhibition of Oct4 promoter activation as a result of p53 binding.
In addition to potentially promoting differentiation of damaged ESCs, p53 has been suggested to prevent the differentiation of neighboring ESCs. A chromatin immunoprecipitation microarray screen carried out using immunoprecipitated p53 protein from mESCs treated with doxorubicin or left untreated, demonstrated a statistically significant increase in binding of p53 to Wnt ligand gene promoters in the treated samples. 37 This binding was specific to the undifferentiated ESCs, since mEFs or ESCs that were differentiated into neural progenitors (NPs) did not display p53 binding to Wnt promoters after doxorubicin treatment. Since Wnt ligands have recently been shown to prevent the differentiation of mESCs over the short term, 38,39 it may be that the damaged ESCs secrete Wnt ligands to prevent the differentiation of neighboring and potentially undamaged cells. In addition, the Nanog protein was downregulated in a p53-dependent manner in the treated cells, substantiating previous reports. However, the authors showed that the production of the Wnt ligands and subsequent signaling was not the result of a compensatory mechanism resulting from Nanog downregulation, but was rather an independent event. To test whether p53 induction of Wnt ligands after DNA damage results in secretion of antidifferentiation factors into the medium, unchallenged ESCs were placed into culture medium derived from cells that had been subjected to UV irradiation. Specifically, when medium from UV treated cells was collected and added to untreated ESCs grown in the absence of leukemia inhibitory factor (LIF), these cells displayed an increased rate of proliferation and a decreased differentiation compared with cells receiving medium from untreated cells. When p53 was diminished with siRNA or when inhibitors of Wnt ligand binding to receptors were utilized, the effect on antidifferentiation was greatly reduced. The authors concluded that p53 can serve two opposing functions in ESCs. On the one hand, it can induce differentiation and/or promote cell death in damaged ESCs to remove them from the stem cell pool and thereby prevent the accumulation of mutations in the population. On the other hand, it can promote cell proliferation and prevent differentiation in undamaged cells, to ensure the entire pool is not eliminated.
DNA damage repair
In addition to maintaining genomic integrity by eliminating cells with damaged DNA from the stem cell population through programmed cell death or differentiation, ESCs also appear to upregulate several DNA damage repair pathways. The remainder of this review focuses on the DNA repair capacities of pluripotent stem cells and compares their similarities and differences to more differentiated cell types.
Base excision repair
Base excision repair (BER) is responsible for the correction of chemically modified DNA bases and the repair of DNA single-strand breaks. 40 Studies that examine BER in ESCs are limited, and none to date have been reported in IPSCs. We have observed that basal levels of proteins involved in BER are significantly elevated in mESCs compared with mEFs. 41 This elevation is consistent with a higher capacity of ESCs than mEFs to repair an oligonucleotide template containing a uracil opposite a guanine in vitro. Another study using hESCs and alkaline comet assays demonstrated that the repair kinetics of hESCs were significantly faster than human fibroblast cell lines (hEFs) or HeLa cells when treated with a high dose of H2O2. 19 It should be noted that the interpretation of data generated from comet assays using mESCs may be confounded due to osmotic shock generating excessive DNA single-strand breaks (which is measured by the comet assay); however, this phenomenon has not been reported to occur in hESCs. 42 In addition, the mRNA levels for several BER genes were also significantly higher in unchallenged hESCs compared with hEFs. 19 However, a measurement of the rate of DNA incision in vitro by OGG1, a DNA glycoslylase involved in BER, using untreated cellular extracts, demonstrated no major differences in OGG1 activity between the hESCs and hEFs. Yet, when hESCs or hEFs were treated with H2O2 and the levels of OGG1 and APE1 proteins were examined, the levels of both proteins were induced to much higher levels after treatment in the hESCs. 19 Available data investigating BER in IPSCs are limited to microarray analysis and quantitative polymerase chain reaction (QPCR), which showed that many genes involved in the BER process display trends similar to that of an hESC line. Both the IPSCs and hESCs generally displayed higher transcript expression compared with the parental lines from which the IPSCs were derived. 21
Double strand break repair
DNA damage in the form of double-strand breaks (DSBs) arises as a result of collapsed DNA replication forks, inhibition of topoisomerases, or reactive intermediates produced by radiation or metabolic byproducts that attack the DNA backbone. 43 Cells have evolved several repair strategies to combat the negative effects resulting from such damage. There are two major pathways to repair DSBs in mammalian cells: homologous recombination (HR) and non-homologous end joining (NHEJ). Repair by HR can be considered a high fidelity form of repair, since the cell utilizes a template consisting of the sister chromatid, homologous chromosome or a homologous sequence to repair the lesion. HR is thought to predominantly occur in the late S and G2 phases of the cell cycle, where sister chromatid templates are more readily available for repair. NHEJ can also repair DNA DSBs faithfully; however, the potential for error-prone repair is great. Unlike HR, NHEJ functions in all phases of the cell cycle. 44 The mechanisms governing the fidelity of NHEJ repair are still not fully understood, but the basic repair mechanism involves the ligation of DNA ends, either directly or after an end processing step. Mutations often result from such processing, since nucleotides may be added or removed prior to the ligation step. NHEJ can also be subdivided into classical NHEJ or backup NHEJ (also known as alternative NHEJ, microhomology-mediated end joining, or MMEJ/MHEJ). The detailed mechanisms of MMEJ have not yet been elucidated.
DSB repair in ESCs has become an area of active study. ESCs must repair DSBs both quickly, due to their abbreviated cell cycling times in vivo, as well as accurately, to prevent the accumulation of mutations. To address the first point, alkaline comet assays have been used to quantitate the DSB repair kinetics in these cells. After exposure to 5 Gy IR, hESCs repaired about 60% of DSBs within one hour, whereas only 15–25% of lesions were repaired by primary human fibroblast lines or HeLa cells during the same time period. 19 After three hours, the number of lesions remaining was similar for most of the cell types. We have observed similar repair kinetics using neutral comet assays, which measure only DNA DSBs, with mESCs and mEFs after treatment with etoposide (Figure 3). Comparisons of the repair kinetics between mESCs and NIH 3T3 cells using fluorescence detected DNA alkaline unwinding assays also demonstrated significantly faster repair rates in the mESCs after IR exposures ranging from 8 to 20 Gy. 45 However, another study that utilized neutral comet assays on mESCs exposed to 75 Gy X-rays has demonstrated an inability to complete repair of approximately 40% of DSBs incurred, even after allowing three hours to repair these lesions. 46 MEFs, as well as hESCs exposed to the same treatment, were capable of repairing 90% of DSBs during the same period. The disparities between these reports have yet to be resolved.

Murine embryonic stem cells (MESCs) repair etoposide-induced double-strand breaks (DSBs) more quickly than fibroblasts. MESCs, NIH 3T3 cells and passage 3 mouse embryo fibroblasts (MEFs) were treated (T) with 3 μmol/L etoposide for two hours and allowed to recover (R) for one to three hours. A neutral pH comet assay was completed and the lengths of comet tails were measured in pixels. All cells were normalized to their respective untreated control. Error bars represent the SEM
It is not surprising that ESCs repair DSBs more frequently using the high fidelity HR pathway rather than the error-prone NHEJ. This contention is supported by the fact that ESCs retain their pluripotential capacity and their ability to produce a complete organism in vivo. Further support derives from the greater availability of homologous templates that can be used as substrates in ESCs compared with mEFs, given the ESC cell cycle dynamic. Lastly, many mouse knockouts of genes coding for proteins involved in HR are early embryonic lethal. In contrast, many components of the NHEJ machinery, when knocked out, are late embryonic lethal, at a time after ESCs would have already differentiated.
Several studies have investigated the ability of ESCs to repair different types of reporter substrates in different systems, some with conflicting results. Early reports demonstrated that mESCs repair a reporter plasmid by HR 81% of the time when that reporter was linearized with the rare cutting endonuclease ISCE1, and that the remaining 19% of repair events consisted of alternative mechanisms such as NHEJ. 47 When this reporter template was modified to contain RAG recognition sites and digested by introducing constructs into mESCs that encode for the RAG recombinase, which is normally involved in the generation of immunoglobulin diversity, NHEJ became the predominant repair mechanism, with a frequency of 92%. 48 As previously noted, however, the RAG proteins direct repair towards the NHEJ pathway through a poorly understood mechanism. 49 Nonetheless, these data suggest that the error-prone NHEJ pathway can be utilized by mESCs under certain conditions, as demonstrated by a drop in repair by NHEJ repair using the RAG template when mESCs lacking core components of NHEJ were tested.
A more recent study compared HR, NHEJ and MMEJ repair between mESCs and mEFs. 50 In this study, the basal levels of proteins involved in HR were highly elevated in the mESCs compared with mEFs, and this elevation correlated with a higher propensity to repair a cellular reporter plasmid which measures HR. Repair was 2–4-fold higher in the mESCs than in mEFs, depending on the genetic background from which the cells were derived. When several NHEJ proteins were examined between the two cell types, their expression was quite variable. Intriguingly, the protein expression of DNA ligase IV, the rate-limiting protein in NHEJ, 51 was found at a much lower level in the mESCs than in mEFs, which also corresponded to a decreased capacity (3–4-fold) to repair a template that measures only NHEJ activity. In support of this finding, DNA-PKcs, which regulates the stability and activity of DNA ligase IV, is also expressed at lower levels in mESCs than in mEFs. 46 When mESCs were induced to differentiate, the level of DNA ligase IV protein increased, concomitant with an increase in repair of the NHEJ reporter. At the same time, repair by HR decreased. 50 When MMEJ was quantified, its activity was found to be at similar low levels in both mESCs and mEFs. Confirmatory observations that quantified HR and NHEJ activity were recently reported using in vitro repair assays and utilizing nuclear cell extracts from mESCs or murine adult ear fibroblasts. 52 In addition, this group also presented data showing that HR is not limited to the S- and G2-phases of the cell cycle as in somatic cells, but occurs in all phases of the cell cycle in mESCs.
DSB repair pathway choice was also recently investigated in hESCs. An undifferentiated hESC line was compared with the same line that was induced to differentiate into NPs or astrocytes with respect to the formation and duration of Rad51 foci after treatment with 2 Gy IR. 53 Since Rad51 localizes to sites of DSBs and is a crucial participant in HR, focus formation of this protein served as a surrogate for HR in this study. In hESCs, Rad51 foci incrementally increased after 2 Gy IR treatment, reaching a peak at six hours postirradiation, at which time more than 75% of cells were positively stained. The NPs reached a peak at 12 h after treatment with about 70% of cells staining positive. Astrocytes showed no Rad51 focus formation after IR. This finding correlates well with elevated basal expression of Rad51 in the hESCs compared with the NPs, which expressed Rad51 protein at a 50% lower level, and with astrocytes, which expressed about 10% of Rad51 protein compared with the hESCs. This finding suggests that hESCs utilize HR to repair DSBs at high frequency, while more differentiated cell types require either more time to activate the HR process, or fail to activate it at all. The data in this study and the initial findings in hESCs DSB repair kinetics by comet assay are somewhat contradictory and this discrepancy has not been addressed.
A study measuring NHEJ activity was also performed using the same cell types as that described above. Using a lentiviral fluorescent-based NHEJ reporter, Adams et al. demonstrated that hESCs were capable of completing NHEJ at a level similar to the NPs. But this repair was observed at much lower levels (2.6-fold lower) than was found in astrocytes after all cell types were allowed to repair the reporter template for 48 h. 54 Consistent with these data, astrocytes derived from hESCs were previously demonstrated to repair IR-induced DSBs in a predominantly DNA-PKcs, and thus in a NHEJ-dependent manner. 53 Over a longer time course of 72 h, the degree of NHEJ repair in hESCs and NPs increased proportionally, resulting in only a 1.6-fold lower repair capacity compared with the astrocytes. Although NHEJ activity was found at lower levels in the hESCs, the accuracy of repair using the NHEJ reporter template was 1.4-fold higher in the hESCs compared with NPs and 2.6-fold higher compared with astrocytes.
DSB repair activity in IPS cells has not been measured. Available data from microarray and subsequent QPCR analysis have shown a significant elevation of several DSB repair transcripts from both major pathways of repair compared with the parental lines from which the IPSCs were derived. 21 This elevation was similar to the transcript expression observed in hESCs. 19
Mismatch repair
Mismatch repair (MMR) is responsible for the repair of a DNA sequence containing errors derived from replication, such as mismatched nucleotides or short stretches of nucleotide insertions/deletions resulting from DNA polymerase slippage. MMR plays a key role in the maintenance of mESC genomes based on the analysis of mutation frequencies in cells proficient or deficient for components of the MMR pathway. As an example, the frequency of spontaneous mutation increased from ∼10−6 in wild-type mESCs cells to ∼10−4 in mESCs lacking MSH2, a critical recognition component of the MMR pathway. 55 Complementing data were recently published demonstrating a similar increase in the spontaneous mutation frequency of mESCs when MSH2 was knocked down with siRNA as well as when an MSH2 mutant mESC line was studied. 56 Similarly, when MMR proficient and deficient mESCs were treated with UV radiation, the induced mutation frequency was significantly higher in the MMR-deficient cells. 55
Consistent with these data, we have found that many proteins involved in the MMR pathway are expressed at high levels under basal conditions in mESCs when compared with mEFs, as were several mRNA transcripts encoding these proteins. 41 To measure MMR activity in mESCs or differentiated mEFs, we employed a plasmid-based fluorescent reporter containing a mismatch that has been previously described. 57 Using this system on unchallenged cells to quantify the inherent ability to repair lesions via the MMR pathway, the mESCs displayed about a 15-fold higher MMR activity over mEFs. 41
Another study has characterized the importance of MutSα, which is comprised of a heterodimer of Msh2 and Msh6, in the apoptotic response in mESCs. A comparison of mESCs with 3T3 cells after treatment with the methylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) showed that mESCs were more sensitive to MNNG than 3T3 cells, even though both cell types had similar levels of O 6 -methylguanine DNA methyltransferase enzymatic activity (MGMT). 31 In addition, chemically inhibiting MGMT increased the sensitivity of mESCs to MNNG treatment and induced significantly more cell death. In contrast, 3T3 cells displayed only a marginal sensitivity and apoptotic phenotype under the same conditions. Since MutSα is also capable of binding O 6 -methylguanine lesions, the authors investigated the role that the MMR pathway may play in the cell death observed in the mESCs. Levels of Msh2 and Msh6 proteins were very highly expressed in mESCs and much lower in 3T3 cells. These proteins also became less abundant in mESCs when they were induced to differentiate. When Msh2 protein was exogenously overexpressed in 3T3 cells, the sensitivity to MNNG increased, especially when MGMT was inhibited. This observation was similar to that observed in undifferentiated mESCs, but in contrast to that observed in differentiated mESCs. The differentiated mESCs were more resistant to killing, suggesting that the expression level of these proteins may in fact play a role in cell death following treatment with MNNG. The elevated level of MutSα in mESCs also correlated with lower expression of the antiapoptotic protein BCL2 under basal conditions, as well as with increased expression of proapoptotic proteins FAS-R, caspases-3 and -7, and BAX after MNNG exposure. These findings were specific to the mESCs and the expression patterns were reversed in 3T3 cells.
To date, there are no rigorous reports of MMR activity and function in hESCs and IPSCs. Recently, Seriola et al. 58 demonstrated that a hESC line derived from a Huntington disease-affected embryo expressed statistically significant elevated levels of MSH2, MSH3, MLH1, PMS2 and MSH6 transcripts and proteins compared with the same cells that were collected at different points during differentiation. A similar trend in MMR gene expression was observed by microarray analysis and subsequent QPCR comparing wild-type hESCs and IPSCs with fibroblasts or parental cell lines. 21
Nucleotide excision repair
Nucleotide excision repair (NER) is active in repairing DNA lesions that occur as bulky adducts, particularly DNA damage resulting from UV radiation damage, such as cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PP). Mouse ESCs are extremely sensitive to UV radiation, as treatment with 2 J/m2 UV produces cell death at a level equivalent to that seen in mEFs treated with 4 J/m2. 59 The hypersensitivity of mESCs to UV and subsequent cell death is not the result of defective DNA repair, since transcription-coupled repair, a subpathway of NER, is functional in wild-type cells. This was demonstrated using mutation frequency assays at the Hprt locus in wild-type mESCs or mESCs that were deficient for components of the NER machinery. 59 Mutant mESCs treated with different dosages of UV displayed mutation frequencies that ranged from one- to four-fold higher than wild-type cells processed in the same manner.
Another study, again using mESCs, examined the repair capacities of these cells, both unperturbed and following UV exposure, by observing the rate of CPDs or 6-4PP removal. 30 To this end, mESCs were treated with doses of 20 J/m2 for analysis of CPDs or 60 J/m2 for analysis of 6-4PPs, after which cells were allowed up to 12 h to repair lesions. The dosages used were those required to ensure that an average of one CPD or 6-4PP, respectively, would be present every 15 kb of DNA. Repair of CPDs was undetectable in these cells, while only 30% of 6-4PPs were repaired by four hours. No further repair of CPDs was observed between 4 and 12 h. To determine if repair was affected by the dose of treatment, mESCs were exposed to increasing doses of UV radiation, and the efficacy of repair was quantified by a DNA replication assay using tritiated thymidine. MEFs treated in a similar manner served as a control. It is important to note that in parallel experiments the UV doses applied to mESCs corresponded to twice the dose used for mEFs to ensure that an equivalent number of lesions were generated in both cell types. The possibility that ESCs repair UV-induced lesions at a faster rate than mEFs, and thus require a higher dose of UV to create an equivalent number of lesions compared with mEFs treated with lower UV doses was not examined. Using this system, mESC repair became saturated and leveled off at an effective dose of 5 J/m2. At this dose and up to 15 J/m2, the mEFs still repaired lesions in a linear fashion. Although the ability of mESCs to repair UV-induced lesions was reduced compared with mEFs, the same study showed that NER plays an important role in preventing the accumulation of mutations in mESCs, particularly at the lower doses. This was demonstrated when wild-type mESCs and mESCs derived from ERCC1 knockout mice were compared following UV irradiation. The ESCs from knockout mice were more sensitive to treatment than were wild-type cells at lower UV doses, and they also displayed a 10-fold higher mutation frequency at Hprt.
NER activity has also been quantified in hESCs but not yet reported for IPSCs. The responses of two hESC lines to damage induced by UVC radiation showed more rapid repair kinetics than two hEF lines when compared by the alkaline comet assay. 19 Basal levels of transcripts coding for NER proteins were not significantly different between the hESCs and hEFs, suggesting that the increased rate of repair was not due to an overabundance of mRNAs encoding NER proteins and subsequent translation of these messages as reported for other repair pathways previously described. Similar trends for message levels have also been observed in IPSCs when they were compared with hESCs and their differentiated cell types. 21
Oxidative stress response
Cells are continuously exposed to reactive oxygen species (ROS), produced from either endogenous sources, such as intermediates and byproducts of oxidative phosphorylation or from exogenous sources, such as free radical production resulting from exposure to different types of radiation. Both ESCs and IPSCs have evolved unique mechanisms to prevent and counteract DNA damage incurred as a result of ROS. 45 When the total levels of ROS were examined in mESCs, mEFs or NIH 3T3 cells, the amount of ROS was higher in the differentiated cell types. When mESCs were induced to differentiate toward a hematopoetic lineage, the level of ROS also increased and was similar to that seen in the mEFs and 3T3 cells. 45 In addition, when mESCs were cultured under hyperoxic (40% oxygen) conditions, they were able to continue proliferating, while mEFs senesced under the same conditions. 45 These observations suggest that mESCs employ one or more mechanisms to maintain low ROS levels. Microarray analysis has demonstrated that undifferentiated mESCs have higher transcript levels of antioxidant genes compared with mESCs that have been induced to differentiate. 45 It has also been suggested that ROS levels can act as switches to induce differentiation in ESCs, as increased levels of free radicals have been reported in ESCs induced to differentiate into a mesoendodermal lineage. 60 We have observed that, like many tumor cell lines, undifferentiated mESCs utilize glycolysis to a greater extent for ATP production than their differentiated counterparts, potentially limiting endogenous ROS production generated from oxidative phosphorylation.
Similar to mESCs, undifferentiated hESCs display decreased levels of total ROS compared with spontaneously differentiated hESCs, as well as lower levels of peroxides and superoxides in the mitochondrial matrix. 61 Antioxidant gene expression is also significantly downregulated as hESCs become differentiated. 61 Consistent with these data, another study demonstrated that hESCs also display lower levels of the DNA adduct 8-oxyguanine in unchallenged cells compared with somatic cell types. 19 It remains unknown, however, whether the results seen in hESCs are due to a reduced ability to produce ROS because they have fewer mitochondria than differentiated cells, and/or whether the elevated level of free-radical scavengers is sufficient to exert a protective effect. 62 A recent investigation into the stress defense capabilities of IPSCs demonstrated that these cells are similar to hESCs when ROS levels and antioxidant gene expression were profiled and compared with the differentiated parental line. 63 Thus, pluripotent embryonic and embryonic-like cells usually display low levels of ROS, which can be preventive of induction of basal oxidative DNA damage.
Conclusion
The accumulation of mutations in a differentiated cell genome may result in the loss or gain of gene functions that are restricted to the particular cell affected, and may contribute to disease at a later time. In contrast, mutations in pluripotent ESCs will be propagated and arise in multiple differentiated cell types of the organism. Studies on mESCs have shown that mutations occur at a much lower frequency than that in differentiated cells. Similar data have not been described for hESCs, but one might assume that a similar finding may apply to these cells, since ESCs derived from both species have similar fundamental characteristics and have the potential to give rise to complete organisms. The mutation frequency in IPSCs, on the other hand, remains enigmatic since reprogramming may or may not result in the activation of mechanisms that prevent the accumulation of mutations in ESCs. These IPSCs, however, will have accumulated mutations during their life as somatic cells, which should cause pause when one considers their use for therapeutic purposes. In particular, the success of generating IPSCs is quite low, and the success rate increases several-fold when p53 is mutated or lost. 64,65 Thus, therapeutic use of IPSCs can result in the expansion of pre-existing mutations and can potentially have detrimental effects, such as tumor formation, in a patient with the passage of time.
The mechanisms by which mutations are suppressed in mESCs, hESCs and potentially IPSCs are several-fold. These cells are hypersensitive to DNA damage and can undergo cell death, thereby eliminating cells with DNA damage from the population after exogenous challenge. Alternatively, they may be induced to differentiate under certain stress, which also eliminates cells with damaged DNA from the self-renewing stem cell pool. A third documented mechanism involves the upregulation of DNA repair pathways to repair damage as quickly as it is identified, a strategy that is not mutually exclusive with the above-mentioned mechanisms. Although the death or differentiation described for these cells is a consequence of an exogenous challenge, the DNA repair pathways are equally or more critical for repairing endogenously derived DNA damage, that would typically occur during normal embryonic development. Consistent with this hypothesis, ESCs and also IPSCs appear to protect against oxidative DNA damage by upregulating genes whose products have antioxidant activity and by supporting a reduced number of mitochondria, which effectively limits the potential to generate free radicals as a consequence of oxidative phosphorylation. Thus, ESCs have evolved multiple unique mechanisms to respond to DNA damage that are significantly different than those utilized by differentiated cell types. These mechanisms are critical not only for the proper development of the embryo but also for the continued survival of future generations. If mutations were to arise in the soma, then the resulting impact could be detrimental to the genetic viability of a species when propagated through several generations. Consistent with species viability, germ cells that both create and derive from ESCs display a lower mutational burden than somatic cells, even as a function of age. 66,67
Although there are similarities between IPSCs and ESCs, differences also exist. Based on available data, IPSCs and ESCs both have the capacity to form all three germ layers upon differentiation. On the other hand, IPSCs initially retain an epigenetic memory reflective of the somatic cell type from which they are derived. 68,69 It should be noted that the epigenetic differences between IPSCs and ESCs can be reduced or even possibly eliminated by extended cell culture or the addition of histone deacetylase inhibitors through a mechanism that is still not understood. 69,70 Additionally, several groups have demonstrated differences in the expression levels of several mRNAs and miRNAs when ESCs and IPSCs were compared. 71–73 Thus, the pluripotent potential of IPSCs may be restricted after extended culture due both to the mutations incurred as somatic cells prior to reprogramming as well as to any transcriptome differences and epigenetic footprinting that IPSCs may retain. The application of using IPSCS for tissue replacement therapy may have limitations, given the need to expand the cells in culture and properly differentiate them into the desired cell type. The generation of IPSCs from somatic cells can also involve the expression of a c-Myc transgene, which, if constitutively overexpressed, elevates the risks for abnormal cell growth and tumorigenesis. Thus, the risk of developing tumors derived from donor cells may be sufficiently high to offset the benefits of IPSC-mediated therapeutics, at least when generated using the c-Myc oncogene. Unless conditions that eliminate such enhanced risks are established, the use of IPSCs may be better suited to in vitro studies, such as individualized approaches to identify patient-specific drugs for the treatment of disease, rather than a tool for regenerative or therapeutic medicine. Given the current limitations of IPSCs, it would appear that ESCs or early adult stem cells are better suited to use for tissue regeneration and repair therapies in the treatment of disease or injury.
Footnotes
Acknowledgements
I thank B Russell for help in editing this manuscript. I also thank P Hexley, G Babcock and the Shriner's Hospital for Children in Cincinnati for assistance with flow cytometry. This work was supported in part by NIH grants R01 ES012695 and T32 ES007250.