
Abstract
Liver fibrosis represents a process of healing and scarring in response to chronic liver injury. Following injury, an acute inflammation response takes place resulting in moderate cell necrosis and extracellular matrix damage. Melittin, the major bioactive component in the venom of honey bee Apis mellifera, is a 26-residue amphipathic peptide with well-known cytolytic, antimicrobial and proinflammatory properties. However, the molecular mechanisms responsible for the anti-inflammatory activity of melittin have not been elucidated in liver fibrosis. We investigated whether melittin ameliorates liver inflammation and fibrosis in thioacetamide (TAA)-induced liver fibrosis. Two groups of mice were treated with TAA (200 mg/L, in drinking water), one of the groups of mice was co-treated with melittin (0.1 mg/kg) for 12 weeks while the other was not. Hepatic stellate cells (HSCs) were cultured with tumor necrosis factor α in the absence or presence of melittin. Melittin suppresses the expression of proinflammatory cytokines through the nuclear factor (NF)-κB signaling pathway. Moreover, melittin reduces the activity of HSCs in vitro, and decreases the expression of fibrotic gene responses in TAA-induced liver fibrosis. Taken together, melittin prevents TAA-induced liver fibrosis by inhibiting liver inflammation and fibrosis, the mechanism of which is the interruption of the NF-κB signaling pathway. These results suggest that melittin could be an effective agent for preventing liver fibrosis.
Introduction
Liver fibrosis is the result of chronic liver injury from multiple causes ranging from viral infections and drug abuse to autoimmune disease, non-alcoholic fatty liver disease and developmental anomalies. 1,2 The processes of liver repair and of fibrogenesis resemble that of a wound-healing process. Viral infection, alcoholic or drug toxicity, or any other factors that cause damage to hepatocytes elicit an inflammatory reaction in the liver. Following injury, an acute inflammation response takes place resulting in moderate cell necrosis and extracellular matrix damage. 3 Hepatic stellate cells (HSCs) are perisinusoidal cells residing in the space of Disse. During injury, in response to inflammatory and other stimuli, these cells adopt a myofibroblast-like phenotype and represent the cornerstone of the fibrotic response in the liver. 4–7 Once activated, HSCs will up-regulate gene expression of extracellular matrix (ECM) components, matrix-degrading enzymes and their respective inhibitors, resulting in matrix remodeling and accumulation at sites with abundant activated HSCs. 8,9 The most effective way to treat liver fibrosis during the early stages is to remove the causative agent. Treatments that are currently under evaluation mainly inhibit HSC activation and proliferation or the release of cytokines and only a few drugs have direct effect on collagen synthesis or degradation. 1,8,10 Although new therapeutic approaches have been recently proposed, there is still no established therapy for liver fibrosis. 11
Bee venom is known to be a very complex mixture of active peptides (e.g. melittin, apamin, adolapin, mast cell degranulation [MCD] peptide), enzymes (e.g. phospholipase A2) and non-peptide component amines. 12,13 Melittin, the major bioactive component in the venom honey bee Apis mellifera, is a 26-residue amphipathic peptide with well-known cytolytic, antimicrobial and proinflammatory properties. 12,14 Although adolapin and MCD peptide have anti-inflammatory activities, these substances are present in very small quantities in the whole bee venom. 15–17 It has been reported that bee venom and melittin play an important role in inflammation. 18 Also, melittin inhibits the enzymatic activity of phospholipase A2, an inflammatory trigger. 19 Although the proinflammatory effect of melittin has been known in activated macrophages, no reports have yet demonstrated a preventive action of HSC regulation or liver fibrogenesis by melittin; moreover, the mechanisms underlying its beneficial effects remain to be elucidated.
It is well known that following long-term feeding of thioacetamide (TAA), rodents can develop liver fibrosis and cirrhosis; these changes closely resemble the clinical and pathophysiological changes seen in humans. 20–22 In the present study, we examined the effect of melittin on TAA-induced murine liver fibrosis and activated HSCs. The aim of the present study was to investigate the therapeutic effects of melittin in liver fibrosis, and to explore its probable mechanisms.
Materials and methods
Animals and TAA-induced liver damage
Eight-week-old (20–22 g, n = 7 for each group) C57BL/6 mice (Orient Bio, Seongnam, Korea) were housed in a room for 12 weeks at 22 ± 2°C and on a 12-h light–dark cycle. The animal experiments were performed in accordance with the NIH guidelines for the care and use of laboratory animals. Liver fibrosis was induced by TAA in eight-week-old C57BL6 mice. TAA (200 mg/L; Sigma-Aldrich, Munich, Germany) were orally administered in drinking water for 12 weeks. C57BL/6 mice were randomly divided into three groups: a normal group without any treatment administered (normal control, NC); a TAA-induced liver damage (TAA) group; and a TAA-induced liver damage treated with melittin (TAA + Mel) group (n = 7, each group). The TAA + Mel group received intraperitoneal injection of melittin (0.1 mg/kg) dissolved in saline twice a week. Mice were sacrificed after 12 weeks from the first TAA administration.
Cell culture and materials
Primary cultured rat HSCs were obtained from Dr Jeong (Kyungpook National University, Daegu, Korea). HSCs were cultivated at 37°C in an atmosphere of 5% CO2 in minimum essential medium, supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 mg/mL streptomycin (Gibco, Carlsbad, CA, USA). Melittin was purchased from Sigma Chemicals Co (St Louis, MO, USA) and tumor necrosis factor (TNF)-α was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Histopathology and immunohistochemistry
Small pieces of liver from each lobe were kept in 10% formalin solution. Paraffin blocks were prepared. Cross-sections taken from the blocks were stained with Masson's trichrome and hematoxylin & eosin stains (H&E). For immunohistochemistry, sections were incubated with anti-TNF-α, anti-fibronectin, anti-interleukin (IL)-6 (Abcam, Cambridge, MA, USA), anti-IL-1β (Santa Cruz Biotechnology), anti-transforming growth factor (TGF)-β1 (R&D Systems, Minneapolis, MN, USA) and anti-α-smooth muscle actin (SMA; Sigma). After three serial washes with phosphate-buffered saline, the sections were processed by an indirect immunoperoxidase technique using a commercial kit (LSAB kit; Dako, Glostrup, Denmark). Light microscopy was used to acquire the immunohistochemical images.
Enzyme-linked immunosorbent assay
Serum TNF-α concentration was measured with a solid phase sandwich enzyme-linked immunosorbent assay (ELISA) using a Quantikine mouse TNF-α kit (R&D Systems). Serum IL-1β concentration was also measured with a solid phase sandwich ELISA using a Quantikine mouse IL-1β kit (R&D Systems).
Western blotting
Cells and tissues were lysed in buffer (50 mmol/L Tris pH 8.0, 150 mmol/L NaCl, 5 mmol/L EDTA, 0.5% NP-40, 100 mmol/L PMSF, 1 mol/L DTT, 10 mg/mL leupeptin and aprotinin; all from Sigma) and centrifuged at 12,000 rpm for 30 min after storing for 30 min on ice. Protein concentration was measured by the Bradford protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Sodium dodecyl sulfate polyacrylamide gel electrophoresis was performed with 8–12% polyacrylamide gels at 100 V for three hours. The resolved proteins were transferred from the gel onto a polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA, USA) and probed with anti-TNF-α, anti-fibronectin, anti-IL-6 (Abcam), anti-IL-1β, anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology), anti-TGF-β1 (R&D Systems), anti-α-SMA (Sigma), anti-p-I-κB kinase (IKK), anti-p-IκB and anti-nuclear factor (NF)-κB (Cell Signaling, Danvers, MA, USA) followed by secondary antibody conjugated to horseradish peroxidase (1:2000) and detected with enhanced chemiluminescence reagents (Amersham Bioscience, Piscataway, NJ, UK). Signal intensity was quantified by image analyzer (Las3000; Fuji, Tokyo, Japan).
Nuclear and cytosolic extract preparation
For cytosolic fractions, liver tissue was homogenized in extraction buffer (10 mmol/L HEPES pH 8.0, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L DTT, 300 mmol/L sucrose, 0.1% NP-40 and 0.5 mmol/L PMSF) for 15 min on ice and centrifuged 6000
DNA binding activity of NF-κB
Electrophoretic mobility shift assay (EMSA) was performed according to the manufacturer's recommendations (A DIG Gel Shift kit; Roche, Mannheim, Germany). The NF-κB oligodeoxynucleotide (ODN) probe (NF-κB: 5′ CTT GAA
Statistical analysis
All values are expressed as means ± standard errors of the mean. Statistical differences in the average between the two groups and among three or more groups were assessed by unpaired t-test and analysis of variance, respectively. All experiments were performed at least three times. P < 0.05 was considered statistically significant.
Results
Histological examination of the livers of TAA-treated mice with or without treatment with melittin
The morphological changes of liver injury and fibrosis caused by TAA were visualized in the sections stained by H&E and trichrome. Histological changes are shown in Figures 1a and b. The control livers showed a normal lobular architecture with central veins and radiating hepatic cords. In the TAA group, the increase and expansion of fibrous septae, ballooning changes of hepatocytes and multifocal hepatocellular necrosis were remarkable. Collagen fibers were distinctly deposited in the TAA group and were visualized by trichrome staining. In contrast, the TAA + Mel group showed less hepatic cell necrosis and the collagen deposition was decreased.
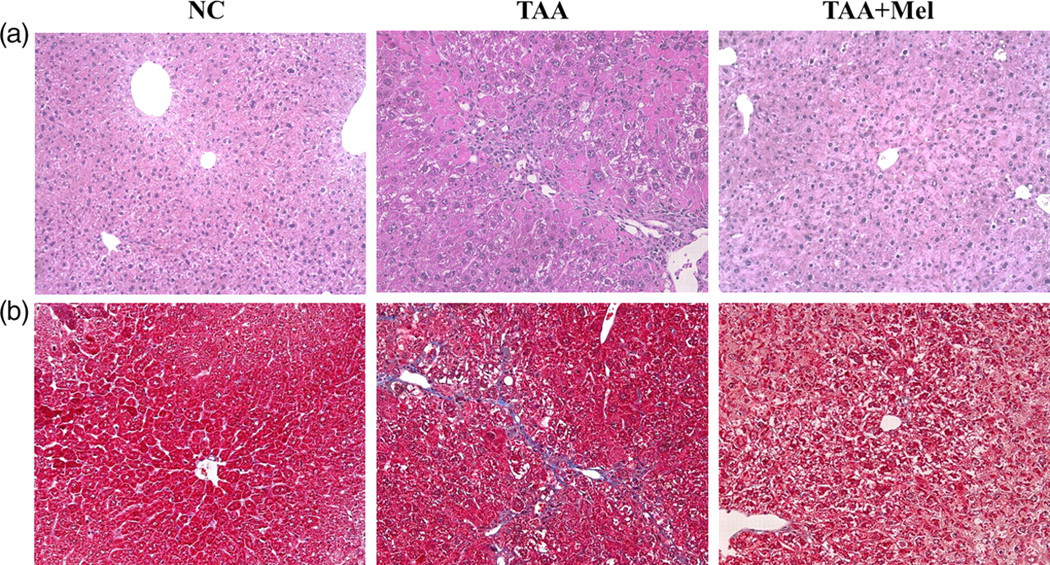
Effect of melittin in thioacetamide (TAA)-induced liver fibrosis. Mice (n = 7) were killed at 12 weeks after TAA administration with or without treatment with melittin. Histological changes in the liver tissues were shown by hematoxylin and eosin (H&E) (a) and trichrome staining (b) (original magnification, ×200). NC, normal control; Mel, melittin. (A color version of this figure is available in the online journal)
Melittin suppressed liver inflammation in the livers of TAA-treated mice
To explore the mechanisms underlying the protective effects of melittin, we proposed that melittin might protect the liver against TAA-induced injury by suppressing inflammation in the liver. The expression of proinflammatory cytokines in the fibrotic livers were evaluated by ELISA, immunohistochemistry and Western blotting. The activities of serum TNF-α and IL-1β were significantly higher in the TAA group than in the NC group, and were significantly lower in the TAA + Mel group than in the TAA group (Figure 2). Immunohistochemistry revealed that vascular cell adhesion protein 1 (VCAM-1), IL-6 and TNF-α were increased after chronic TAA administration (Figure 3a). Treatment with melittin significantly abrogated this activation. Also, the expression levels of VCAM-1, IL-6 and TNF-α were significantly increased in the TAA group. The levels of VCAM-1, IL-6 and TNF-α in the liver were significantly reduced by treatment with melittin (Figure 3b). Collectively, these data indicate that melittin significantly reduced the levels of proinflammatory cytokines after TAA administration, which may result in the inhibition of TAA-induced liver fibrosis.
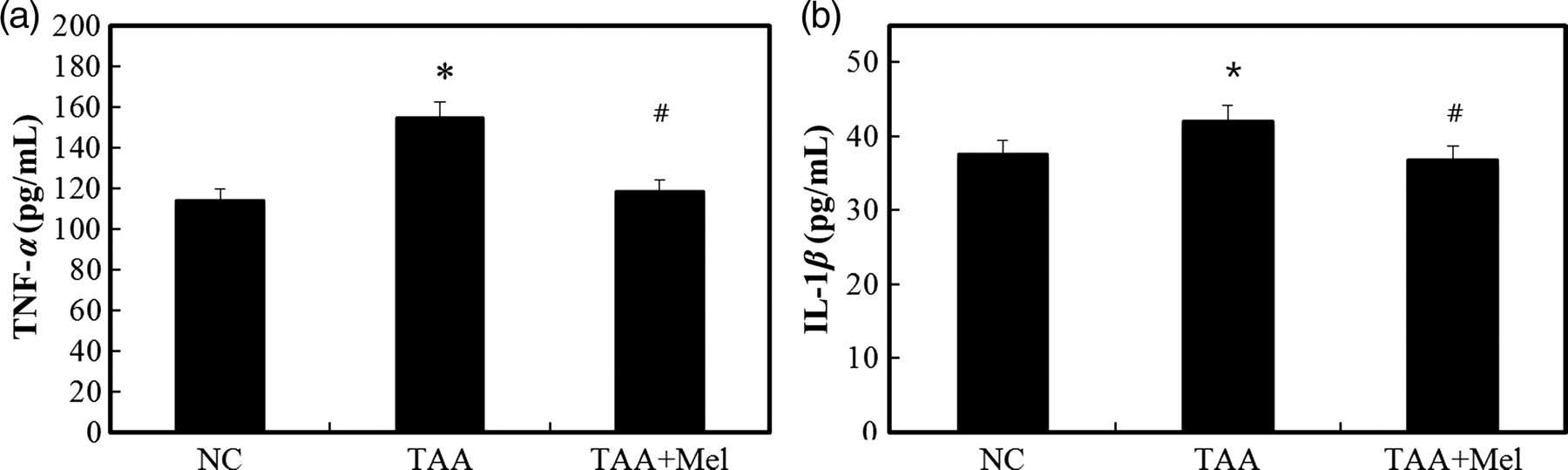
Inhibitory effect of melittin on the expression of proinflammatory cytokines in thioacetamide (TAA)-induced liver fibrosis. Enzyme-linked immunosorbent assay (ELISA) results for tumor necrosis factor (TNF)-α (a) and interleukin (IL)-1β (b) in the serum. Increase in serum concentrations of TNF-α and IL-1β were observed in the TAA group but were suppressed in the TAA + Mel group. Data represent means ± SD of the seven animals in each group. *P < 0.05 versus NC group and # P < 0.05 versus TAA group. NC, normal control; Mel, melittin

Melittin suppresses the expression of proinflammatory cytokines in thioacetamide (TAA)-induced liver fibrosis. (a) Immunostaining for vascular cell adhesion protein 1 (VCAM-1), interleukin (IL)-6 and tumor necrosis factor (TNF)-α in TAA-treated mouse liver (original magnification, ×200). (b) Western blotting for VCAM-1, IL-6 and TNF-α in TAA-treated mouse liver. The results are indicated as the means ± SD of three separate experiments. *P < 0.05 versus NC group and # P < 0.05 versus TAA group. NC, normal control; Mel, melittin (A color version of this figure is available in the online journal)
Melittin suppressed liver fibrosis in the livers of TAA-treated mice
Immunohistochemical experiments were performed to further evaluate the impact of melittin in the regulation of expression of the genes relevant to liver fibrogenesis, including TGF-β1, α-SMA and fibronectin. As shown in Figure 4a, the expression of TGF-β1, α-SMA and fibronectin is hardly detectable in the normal mouse liver. Administration of TAA significantly increased the number of cells positive for TGF-β1, α-SMA and fibronectin. The treatment with melittin significantly reduced the number of cells positive for TGF-β1, α-SMA and fibronectin.
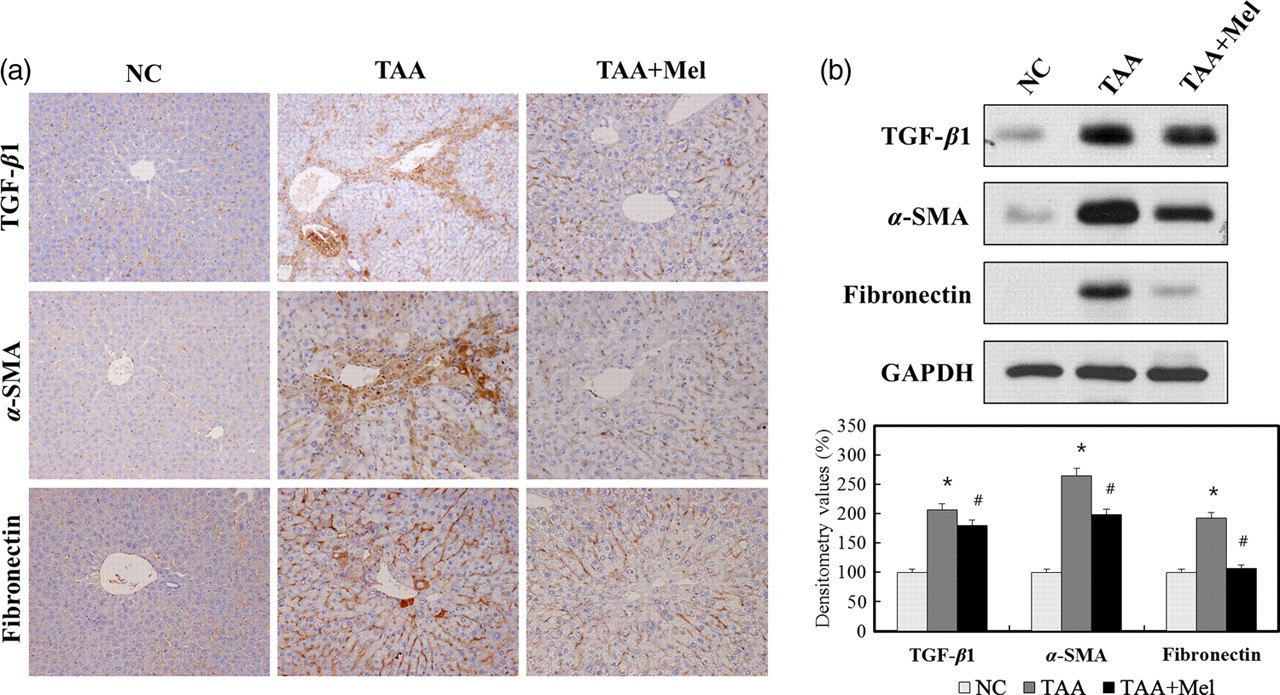
Melittin suppresses the expression of fibrosis-related cytokines in thioacetamide (TAA)-induced liver fibrosis. (a) Immunostaining for transforming growth factor (TGF)-β1, α-smooth muscle actin (α-SMA) and fibronectin in TAA-treated mouse liver (original magnification, ×200). (b) Western blotting for TGF-β1, α-SMA and fibronectin in TAA-treated mouse liver. The results are indicated as the means ± SD of three separate experiments. *P < 0.05 versus NC group and # P < 0.05 versus TAA group. NC, normal control; Mel, melittin (A color version of this figure is available in the online journal)
To investigate the biological activity of melittin in fibrotic liver, TGF-β1, α-SMA and fibronectin were assessed in the liver homogenates by Western blotting. As shown in Figure 4b, the expression of TGF-β1, α-SMA and fibronectin markedly increased in the TAA-treated fibrotic liver. Treatment with melittin significantly abrogated this increase in the TAA + Mel group, which is consistent with the immunohistochemical results. These data collectively suggested that melittin might protect the liver against TAA-induced injury by attenuating fibrotic gene expression.
Melittin suppressed liver fibrosis through the NF-κB signaling pathway
To evaluate the underlying mechanisms of melittin involved in suppression of liver inflammation in vivo by melittin, the expression levels of cytosolic phospho-IKKα, phospho-IκB and nuclear NF-κB were determined. Following TAA administration, there was an increase in cytosolic phospho-IKKα, phospho-IκB and nuclear NF-κB expression in the TAA group. Treatment with melittin significantly reduced the IKKα and IκB phosphorylation in the TAA + Mel group. Treatment with melittin also reduced the amount of the nuclear NF-κB protein (Figure 5a). Also, the inhibitory effect of melittin on transcriptional activity of NF-κB was confirmed by EMSA. Treatment of melittin significantly attenuated NF-κB binding to DNA induced by liver fibrosis (Figure 5b). Taken together, these results indicate that treatment with melittin abrogated the effect of TAA on altering the expression level of genes which are relevant to liver inflammation through NF-κB signaling.
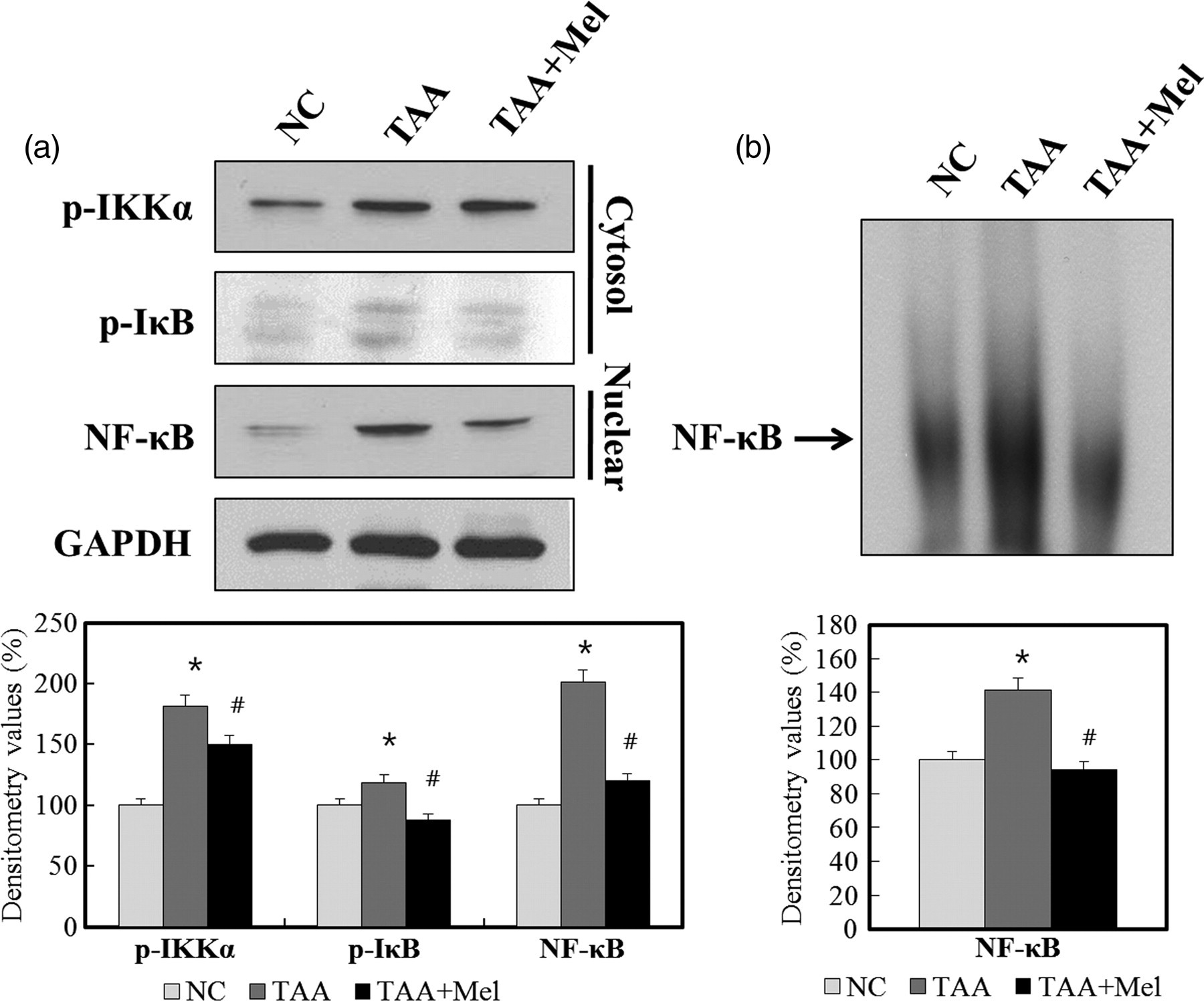
Effect of melittin on nuclear factor (NF)-κB signaling pathway in thioacetamide (TAA)-induced liver fibrosis. (a) Western blotting for phospho-IKKα, phospho-IκB and nuclear NF-κB in TAA-treated mouse liver. Treatment with melittin decreases phosphorylation of IKKα and IκB in cytoplasmic extract of liver tissue. Treatment with melittin also reduces phosphorylation of NF-κB in nuclear extract of liver tissue. (b) Treatment with melittin reduces DNA-binding activity of NF-κB in nuclear extract of liver tissue by electrophoretic mobility shift assay. The results are indicated as the means ± SD of three separate experiments. *P < 0.05 versus NC group and # P < 0.05 versus TAA group
Effect of melittin on inflammation in the TNF-α-treated HSCs
Elevated levels of TNF-α were seen in human and animal models of chronic liver disease. 23,24 Since TNF-α may have a role as an initiator and regulator of the inflammatory response in the fibrotic liver, we tested the effect of melittin following stimulation by TNF-α in HSCs. We assessed the effect of melittin on secretion of the proinflammatory mediators following stimulation by TNF-α. As shown in Figure 6, melittin inhibited the TNF-α secretion in HSCs. Furthermore, melittin also inhibited the TNF-α-induced expression of IL-1β and IL-6, especially with 0.5 μg/mL of melittin.
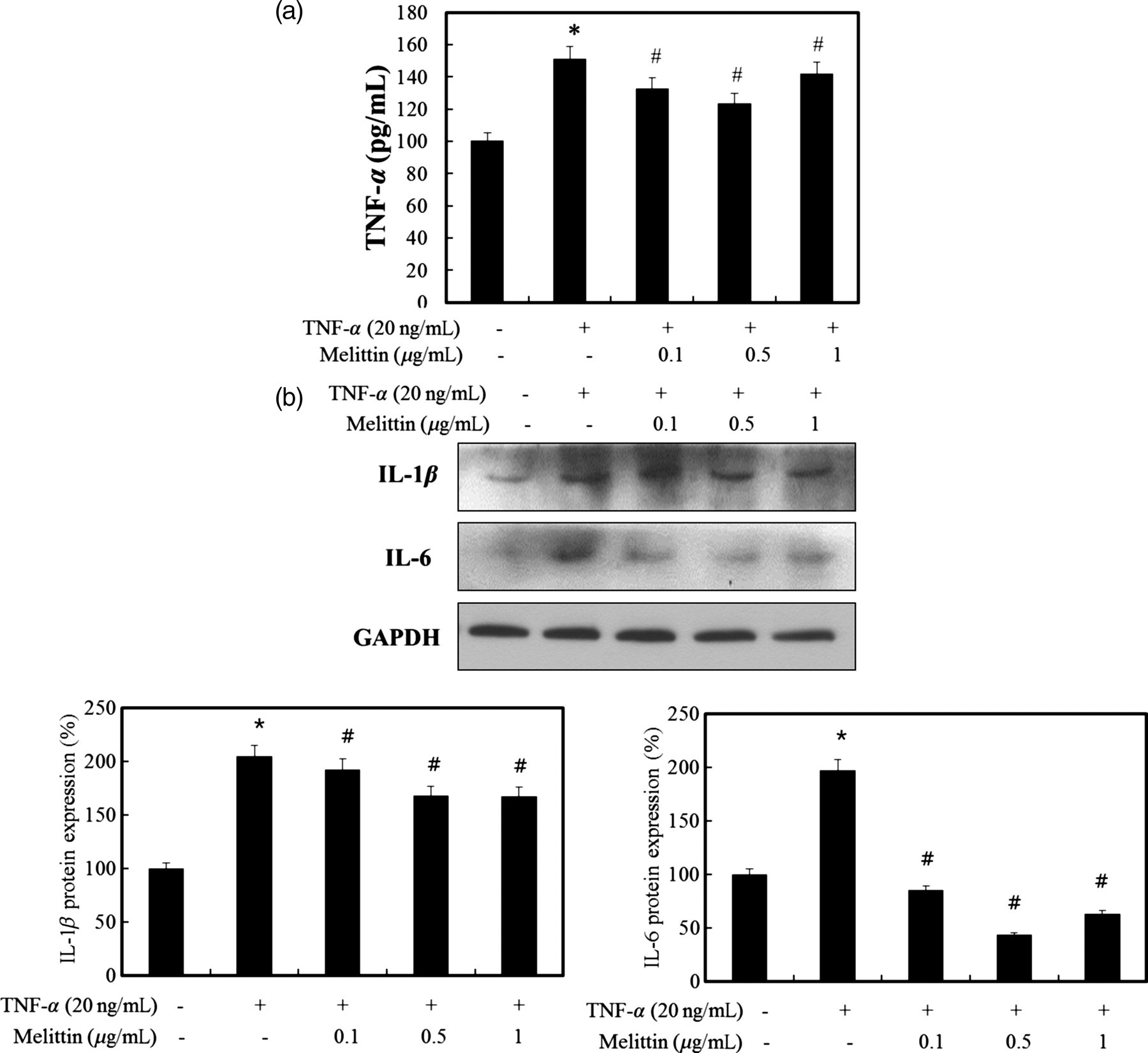
Effect of melittin on inflammatory cytokines in activated hepatic stellate cells (HSCs). (a) Treatment with melittin reduces tumor necrosis factor (TNF)-α cytokine expression induced by TNF-α (20 ng/mL) in HSCs. (b) Melittin effectively inhibits the protein expression of interleukin (IL)-1β and IL-6 induced by TNF-α (20 ng/mL) in HSCs. The results are indicated as the means ± SD of three separate experiments. *P < 0.05 versus normal and # P < 0.05 versus TNF-α treatment group
Discussion
TAA, although not toxic itself, is metabolized into potent hepatotoxins by hepatic cytochromes. These can produce liver injury by the formation of highly reactive compounds and possibly also by activating NF-κB. 25–27 The induction of oxidative stress is a key feature in the destruction of parenchyma and leads to necrosis, fibrosis and cirrhosis. 28 Liver cirrhosis is considered as an irreversible process characterized by excess ECM deposition in the liver with scar formation and destruction of the normal liver architecture. Therefore, resolution of liver fibrosis is a complex process requiring discontinuation of the injurious agent and restoration of the proper balance between liver inflammation and degradation of matrix. 29,30 From this viewpoint, effective treatments for liver fibrosis are earnestly required.
The majority content of the bee venom is melittin, which accounts for 50–60% of the dry weight of bee venom. Melittin has been studied extensively due to its lytic effects on biological and model membranes when inserted into phospholipid layers in high concentrations. 12,31 In contrast, concentrations of melittin lower than 2 μmol/L do not disrupt cell membranes of leukocytes. 32 Several reports suggest that melittin reduces proinflammatory mediators. 33 Although previous studies have demonstrated that bee venom possesses antiproliferative and proapoptotic effects, the precise mechanisms of action of melittin remain to be elucidated. We recently demonstrated that treatment with bee venom provided hepatoprotective effects in TGF-β1-induced hepatocyte apoptosis. 34 We also demonstrated that several factors, including TGF-β1 and TNF-α, produced by activated HSCs or myofibroblasts, were associated with liver fibrosis, and that bee venom could suppress liver fibrosis by inhibiting these factors in a CCl4-induced liver fibrosis model. 35 Following our previous study, we focused our attention especially on the anti-inflammatory and antifibrotic actions of melittin in vivo and in vitro.
In this study, we investigated the effects of melittin in liver fibrosis following chronic injuries by TAA and explored the modes of action of cytokines related with liver inflammation and fibrosis. Inflammation is commonly associated with liver fibrogenesis during chronic liver disease. 36 Proinflammatory cytokines TNF-α and IL-6 are major players in liver inflammation. TNF-α promotes the development of liver fibrosis. 37 IL-6 induces liver inflammation and collagen synthesis in liver fibrosis. Also, cell adhesion molecules are critical for the localization of leukocytes at sites of inflammation and are known as prognostic markers of liver fibrosis. 38 The expressions of VCAM-1, modulated by TNF-α, are up-regulated in alcoholic hepatitis, CCl4-induced liver injury and nutritional fibrosis. 39–41 Moreover, blocking VCAM-1 has been a successful therapeutic strategy to attenuate liver injury, especially the sinusoidal endothelial cell layer. 42–44
Melittin not only suppressed the expression of inflammatory cytokines (TNF-α and IL-6) effectively but also adhesion molecules. These molecules are regulated by NF-κB, a key transcription factor that regulates inflammatory responses, 45,46 which is ordinarily retained in the cytoplasm in an inactive form through association with one of IκB inhibitory proteins. Activation of NF-κB is associated with phosphorylation and subsequent degradation of IκB. The released NF-κB complex translocates to the nucleus and specifically binds to promoter regions of a large number of genes involved in the inflammatory responses, such as those encoding VCAM-1, TNF-α and IL-6. 47,48 To demonstrate NF-κB signaling pathway in liver inflammation, we investigated the translocation of NF-κB protein complex and DNA binding activity of NF-κB. Our results showed that melittin reduces transcriptional activity of NF-κB and effectively inhibits the NF-κB signaling pathway during liver fibrogenesis in vivo.
During liver fibrogenesis, HSCs are activated by inflammatory cytokines and growth factors in a paracrine and autocrine manner. In addition, TGF-β1 is the most potent fibrogenic cytokine described in the activation of HSCs and its expression affects excessive synthesis of ECM components, such as collagen and fibronectin. 49 Down-regulation of TGF-β1 plays a key role in cytokine regulation in liver fibrosis. In this report, melittin significantly reduced the expression levels of TGF-β1. In accordance with the expression of TGF-β1, melittin reduced the expression levels of α-SMA, which is a marker for activated HSCs in liver fibrosis. Moreover, melittin effectively suppressed liver fibrosis and markedly reduced the expression level of fibronectin in the TAA model.
In summary, melittin protected against TAA-induced liver fibrosis by suppressing liver inflammation and fibrogenesis through the NF-κB signaling pathway. In addition, its antifibrotic effect may be attributed to modulation of the inflammatory effect in the activated HSCs. These results confirm and extend our prior observations 34,35 and provide novel insights into the mechanisms of action of melittin in the protection of liver. Our results suggest that melittin might be a therapeutic agent for the treatment of liver fibrosis.
Footnotes
Acknowledgements
This work was supported by a grant from the Research Institute of Medical Science, Catholic University of Daegu (2009) and by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2009-353-E00022).