
Abstract
Heme oxygenase-1 (HO-1) is a cytoprotective enzyme, which regulates cell proliferation and has potential antifibrogenic properties. In the present study, we investigated the effects of pre-emptive HO-1 induction by cobalt protoporphyrin IX on the healing of myocardial infarction in rats. The proliferation and repair of cardiac cells was assessed by immunostaining of Ki67 and proliferating cell nuclear antigen, and apoptosis of cardiomyocytes by terminal deoxynucleotidyl transferase dUTP nick end labelling. Compared with control hearts, HO-1 induction reduced apoptosis and increased proliferation and repair of cardiomyocytes in the infarct border area during the first few days after infarction. Concomitantly, HO-1 decreased accumulation and proliferation of fibroblasts, and down-regulated procollagen type I expression in the infarct area. Furthermore, HO-1 increased expression of the anti-inflammatory cytokine, transforming growth factor-β1, suggesting that the cardioprotective effect of HO-1 in the early phase of infarct healing may result partly from the suppression of the inflammatory response. In the remote myocardium, HO-1 inhibited both proliferation and apoptosis of cardiomyocytes, attenuated heart failure-induced increase in the repair of cardiomyocytes and decreased perivascular fibrosis, thereby potentially alleviating adverse ventricular remodelling. The cardioprotective effects of HO-1 in the late phase of infarct healing may be mediated partly by down-regulation of the profibrotic connective tissue growth factor (CTGF), as HO-1 decreased CTGF expression at week 4. In conclusion, our findings suggest an important role for HO-1 in maintaining cellular homeostasis in the postinfarction heart. Modulation of the HO-1 pathway may provide a new therapeutic approach to enhance the recovery of myocardial infarction and protect against pathological myocardial changes.
Keywords
Introduction
Myocardial infarction (MI) is followed by structural changes both in the infarct area and in the non-infarcted remote myocardium. The process of postinfarction cardiac remodelling is characterized by activation and proliferation of fibroblasts, and formation of fibrous scar in the infarct area along with apoptotic loss of cardiomyocytes that is insufficiently compensated by formation of new cardiomyocytes, and fibrosis and hypertrophy of the non-infarcted myocardium. While the formation of infarct scar is needed to provide mechanical support to the infarcted heart, the adverse cellular and extracellular changes in the infarcted myocardium may eventually lead to the development of heart failure.
Heme oxygenase-1 (HO-1) is a stress-responsive enzyme that catalyzes the degradation of heme into cytoprotective HO-reaction products: biliverdin and bilirubin, carbon monoxide (CO) and free iron. 1–4 HO-1 is also cardioprotective. The beneficial properties of HO-1 have been demonstrated in various cardiovascular disease models in vivo and in vitro. 5,6 There is accumulating evidence that HO-1 promotes angiogenesis. 7 Furthermore, we have recently shown that HO-1 and CO have potential roles in cardiac regeneration after MI. 8 Pre-emptive HO-1 induction promoted angiogenesis and CO-donor pretreatment activated c-kit+ stem/progenitor cells and promoted vasculogenesis and formation of new cardiomyocytes after myocardial infarction in rats. 8 HO-1 gene transfer has earlier been shown to protect against angiotensin II-induced cardiomyocyte apoptosis in vitro, and against ischemia/reperfusion (I/R) injury and cardiomyocyte apoptosis after repeated I/R episodes in rat hearts in vivo. 9,10 In addition, HO-1 induction by hemin has been shown to attenuate left ventricular hypertrophy and fibrosis in adult spontaneously hypertensive rats. 11 Interestingly, HO-1 also regulates the cell cycle in a cell-specific manner: it inhibits proliferation of vascular smooth muscle cells (VSMCs) and fibroblasts, whereas it increases proliferation of endothelial cells. 12–16
As HO-1 may potentially modulate the recovery of the infarcted heart and the left ventricular remodelling process at a number of levels, we studied the temporal and spatial effects of HO-1 on survival and proliferation of cardiac cells, and on fibrosis and expression of major extracellular matrix components during the healing of acute myocardial infarction.
Materials and methods
Experimental MI
Myocardial infarction was produced by permanent ligation of the left anterior descending (LAD) coronary artery as described earlier. 8 The sham-operated rats underwent the same procedure, except for the ligation of the coronary artery. The experimental protocol was approved by the Helsinki University Laboratory Animal Committee and the Provincial State Office of Southern Finland. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). A total of 120 adult male Wistar rats weighing 280–380 g were used in the study. Rats were given a single dose of cobalt protoporphyrin IX (CoPPIX; Frontier Scientific, Logan, UT, USA; 5 mg/kg, intraperitoneally) 24 h prior to LAD ligation to achieve maximal HO-1 induction at the time of LAD ligation (n = 60) as described earlier. 8 Control rats received no pretreatment (n = 60). Rats were anesthetized using medetomidine (Domitor®; Orion, Turku, Finland; 0.5 mg/kg subcutaneously) and ketamine (Ketalar®; Parke Davis, Barcelona, Spain; 60 mg/kg subcutaneously). The heart was exposed through a lateral thoracotomy and the left coronary artery was ligated about 3 mm from its origin. Postoperatively, rats were hydrated with physiological saline subcutaneously and given buprenorphin 0.05 mg/kg subcutaneously twice a day for three days for analgesia (Temgesic®; Schering-Plough, Brussels, Belgium). The mortality rate of infarcted rats was 7% (8 out of 120). Rats were sacrificed one day, three days, one week or four weeks after LAD ligation. The heart was excised, weighed and cut into 2-mm transverse slices below the point where the coronary artery was ligated. The myocardial samples were immediately frozen in liquid nitrogen and stored at −80°C or fixed in 4% neutral buffered formalin for 24 h and embedded in paraffin.
Histology, planimetry and terminal deoxynucleotidyl transferase dUTP nick end labelling
The presence of signs of either acute myocardial infarction (eosinophilia, karyolysis, leukocyte infiltration) or collagen scars compatible with old infarction was analyzed by examination of transverse left ventricular sections stained with Weigert van Gieson. Hearts with no histological signs of infarction were not included in the study (n = 10). Infarct size was determined planimetrically as the ratio of infarcted tissue or scar to the length of the entire left ventricular endocardial circumference as described previously. 17 Apoptosis of cardiomyocytes was determined with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) as described earlier. 18
Realtime quantitative reverse transcription polymerase chain reaction
Infarct size, number of rats included in analyses and HO-1 expression in the infarct border area and in the sham-operated hearts
Values are mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 versus control infarct hearts. † P < 0.05, †† P < 0.01 and ††† P < 0.001 versus control sham hearts
Infarct sizes and the expression of HO-1 in sham-operated hearts have been reported in a previous article with partially the same samples 8
HO-1, heme oxygenase-1
Western blotting and HO-1 enzyme-linked immunosorbent assay
Heart tissue was homogenized as previously described. 8 Equal amounts of protein (30 μg) were fractionated by 6% sodium dodecyl sulfate polyacrylamide gel and blotted onto the nitrocellulose membrane (Hybond-C Extra; Amersham Biosciences, Buckinghamshire, UK). Equal loading was controlled by Ponceau S staining. The membranes were probed first with goat antiprocollagen type I antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Enhanced chemiluminescence detection was performed using the SuperSignal West Pico Chemiluminescent System (Pierce, Rockford, IL, USA). Subsequently, filters were stripped for 20 min at room temperature (0.1 mol/L glycine buffer, pH 2) and re-probed with mouse anti-vimentin (clone V9; NeoMarkers, Fremont, CA, USA) and rabbit anti-actin (Sigma, St Louis, MO, USA) antibodies. Signal intensities were quantified using Gel Doc Image Analyzer (Bio-Rad, Hercules, CA, USA). HO-1 protein was measured using StressXpress HO-1 Elisa kit (Assay Designs, Victoria, Canada) according to kit instructions.
Myocardial and perivascular fibrosis
Five-micrometer transverse paraffin sections from the mid-ventricular level of rat hearts were stained with collagen-specific picrosirius red. Interstitial fibrosis was analyzed from 6–10 randomly selected microscopic fields (×100 or ×200 magnification) as a percentage of the total left ventricular area by using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Perivascular fibrosis was analyzed by calculating the ratio of fibrotic area surrounding the vessel to the total vessel wall area. In all, 6–15 vessels/heart were measured.
Immunohistochemical analysis
Immunofluorescent staining was performed to analyze cardiomyocyte cross-sectional area, total cell number and fibroblast number/mm2, and the proliferation and repair of cells in the rat hearts after MI. Five-micrometer transverse paraffin sections from the mid-ventricular level of rat hearts were stained with the following antibodies: Ki67 (rabbit monoclonal, NeoMarkers), cardiac myosin heavy chain (MHC; mouse monoclonal; Upstate, Temecula, CA, USA), phosphorylated histone H3 (H3P; rabbit polyclonal; Cell Signaling Technology, Danvers, MA, USA), proliferating cell nuclear antigen (PCNA; mouse monoclonal; Cell Signaling Technology), cardiac troponin I (cTnI; rabbit polyclonal; Santa Cruz Biotechnology) and vimentin (mouse monoclonal; clone V9; NeoMarkers). Alexa Fluor 488-conjugated goat anti-mouse IgG (Molecular Probes, Eugene, OR, USA) and TRITC-conjugated swine anti-rabbit IgG (Dako Cytomation, Glostrup, Denmark) were used as secondary antibodies. Nuclei were stained with 4′6-diamino-2-phenylindole (DAPI; Molecular Probes). The immunolabelled sections were examined using a Leica DM 4500B fluorescent microscope (Leica Microsystems GmbH, Wetzlar, Germany).
The cross-sectional area of cardiomyocytes was analyzed from tissue sections stained with anti-MHC and DAPI. Two tissue sections of each heart were analyzed and the cross-sectional area of 100 cardiomyocytes/heart was measured by using ImageJ software. The cardiomyocytes cut in short axis with a visible nucleus were measured.
Two tissue sections of each heart were stained with vimentin and Ki67 antibodies, and DAPI to quantitatively assess the total cell number/mm2, the number of fibroblasts/mm2 and the proliferation of cells. The total cell number/mm2 was assessed by counting the number of DAPI+ nuclei from six randomly selected microscopic fields (×200 or ×400 magnification) photographed from each of the infarct, border and remote areas. The overall proliferation of cells was determined by counting the proportion of Ki67+ nuclei. Fibroblasts were identified by vimentin staining and the characteristic morphology of fibroblasts. The number of vimentin+ fibroblast-like cells/mm2 was counted from six randomly selected microscopic fields (×200 or ×400 magnification) photographed from each of the infarct, border and remote areas. The proliferation of fibroblasts was determined by counting the proportion of vimentin and Ki67 double-positive cells.
Proliferation and repair of cardiomyocytes were analyzed by counting cardiomyocytes positive for proliferation marker Ki67 and marker of DNA synthesis and repair, PCNA. Cardiomyocytes were identified with anti-MHC or anti-cTnI antibody. To quantitatively assess the number of Ki67+ and PCNA+ cardiomyocytes, the proportions of infarct area, border area and remote myocardium of the total area of the ventricular sections and the number of cardiomyocytes/mm2 of border area and remote myocardium were determined. Two tissue sections from each heart were thoroughly examined and all Ki67+ and PCNA+ cardiomyocytes were counted.
Statistical analysis
The results shown are presented as mean ± SEM. Differences between groups were analyzed by Student's t-test or by non-parametric Mann–Whitney U test. A value of P < 0.05 was considered statistically significant.
Results
Infarct size and HO-1 mRNA and protein expression
We found no statistical differences in infarct size, although there was a trend for smaller infarcts in the HO-1-induced group at weeks 1 and 4 after infarction compared with control MI hearts (P = 0.07 and 0.24, respectively) (Table 1).
To confirm the induction of HO-1 by administration of CoPPIX, HO-1 mRNA and protein levels were measured by realtime RT-PCR and HO-1 enzyme-linked immunosorbent assay from the heart tissues of infarcted and sham-operated rats. Administration of a single dose of CoPPIX resulted in powerful and prolonged increases in HO-1 expression as shown earlier. 8,20 HO-1 mRNA increased 2.7-fold in the infarct border areas of HO-1-induced hearts compared with control MI hearts and nine-fold in HO-1-induced, sham-operated hearts compared with control sham hearts at day 1 (P < 0.01) (Table 1). HO-1 mRNA levels declined slowly thereafter, but remained 1.6-fold higher in the infarct border area of HO-1-induced hearts and two-fold higher in sham-operated, HO-1-induced hearts compared with control infarct and sham groups at week 4 (P < 0.01 and P < 0.001, respectively) (Table 1).
HO-1 protein levels were also increased by CoPPIX pretreatment. HO-1 protein levels were two-fold higher in HO-1-induced, sham-operated hearts compared with control sham hearts at day 1 (P < 0.01), whereas in the infarct border area, HO-1 protein levels were increased in both HO-1-induced hearts and control MI hearts (Table 1). At day 3, HO-1 protein levels were about 1.5-fold higher in the infarct border area and three-fold higher in the sham-operated hearts compared with control infarct and sham hearts (P < 0.05 and P < 0.001, respectively) and remained 2.6- and 2.3-fold higher at week 1 (P < 0.05 and P < 0.01, respectively), and 1.4- and 1.5-fold higher at week 4 (P < 0.05) (Table 1).
HO-1 induction inhibits cardiomyocyte apoptosis
Apoptosis of cardiomyocytes was assessed by TUNEL. In sham-operated hearts, the mean level of cardiomyocyte apoptosis was low (0.016 ± 0.0034%) and no differences were found between groups. In the infarcted hearts, HO-1 induction significantly reduced cardiomyocyte apoptosis in the infarct border area at day 3 (HO-1 0.59 ± 0.04% versus control MI 0.91 ± 0.09%, P = 0.006), week 1 (0.16 ± 0.01% versus 0.31 ± 0.05%, P = 0.004) and week 4 after infarction (0.18 ± 0.03% versus 0.31 ± 0.03%, P = 0.008) (Figure 1a). Decreased rates of cardiomyocyte apoptosis were also seen in the remote myocardium at week 1 (HO-1 0.024 ± 0.0047% versus control MI 0.054 ± 0.011%, P = 0.026) and week 4 after MI (0.056 ± 0.006% versus 0.093 ± 0.01%, P = 0.011) (Figure 1b).
HO-1-induction decreased the amount of cardiomyocyte apoptosis. (a) Proportion of TUNEL+ apoptotic cardiomyocytes in the infarct border area, and (b) in the remote myocardium. Values are mean ± SEM. *P < 0.05 and **P < 0.01 versus control group. Number of hearts/group is shown in Table 1. TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; HO-1, heme oxygenase-1
Effect of HO-1 induction on cardiomyocyte proliferation
Immunohistochemical analysis of Ki67 was performed to assess proliferation of cardiomyocytes. In sham-operated hearts, the cardiomyocyte proliferation rate was very low (0.0024 ± 0.0005%) and no differences were found between the groups. In the infarcted hearts, HO-1 induction increased the number of Ki67+ cardiomyocytes three-fold in the infarct border area at day 1 (HO-1 0.37 ± 0.06% versus control 0.12 ± 0.02%, P = 0.004) and two-fold at day 3 (0.46 ± 0.05% versus 0.24 ± 0.04%, P = 0.005) (Figure 2a). However, HO-1 induction had reduced the number of Ki67+ cardiomyocytes in the infarct border area by week 1 after MI (HO-1 0.04 ± 0.01% versus control 0.13 ± 0.02%, P = 0.015; Figure 2a). Similar reductions of Ki67+ cardiomyocytes were also seen in the remote myocardium at day 3 (HO-1 0.0019 ± 0.0001% versus control 0.0053 ± 0.0004%, P = 0.000), week 1 (0.0045 ± 0.0007% versus 0.0151 ± 0.0012%, P = 0.001) and week 4 (0.0084 ± 0.001% versus 0.0142 ± 0.0009%, P = 0.008) (Figure 2b). We also found a few occasional cardiomyocytes positive for the mitosis marker H3P (Figure 2e). However, the number of H3P+ cardiomyocytes was too low to reliably quantify the proportion of mitotic cardiomyocytes in different groups.
Proliferation of cardiomyocytes after myocardial infarction. (a) Proportion of Ki67+ cardiomyocytes in the infarct border area, and (b) in the remote myocardium. Values are mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 versus control group. Number of hearts/group is shown in Table 1. (c) A Ki67+ cardiomyocyte in the infarct border area and (d) in the remote myocardium. (e) Mitotic cardiomyocyte positive for phosphorylated histone H3 (H3P) in the infarct border area. Scale bar = 20 μm. (The number of Ki67+ cardiomyocytes in the infarct border area at week 4 has been reported in an previous article with partially the same samples8). MHC, cardiac myosin
Effect of HO-1 induction on cardiomyocyte DNA repair
The repair of cardiomyocyte DNA was studied by immunohistochemical analysis of PCNA, a marker of both DNA synthesis and repair. In the sham-operated hearts, the number of PCNA+ cardiomyocytes was 0.0077 ± 0.0024% and did not differ by pretreatment or time point. In the infarcted hearts, HO-1 induction increased the number of PCNA+ cardiomyocytes three-fold in the infarct border area at day 1 (HO-1 4.78 ± 0.67% versus control 1.53 ± 0.20%, P = 0.030) and 2.3-fold at day 3 (4.91 ± 0.59% versus 2.10 ± 0.29%, P = 0.009) (Figure 3a). Similar to the Ki67-results, we found a trend for a reduced number of PCNA+ cardiomyocytes in the infarct border area at week 1 (0.43 ± 0.11% versus 0.78 ± 0.13%, P = 0.072) and week 4 after MI (0.69 ± 0.18% versus 1.37 ± 0.37%, P = 0.074) (Figure 3a). In contrast, the number of PCNA+ cardiomyocytes in the remote myocardium was remarkably stable throughout the study in HO-1-induced hearts (0.023 ± 0.005%). In control hearts, the number of PCNA+ cardiomyocytes remained unchanged in the remote myocardium from day 1 to week 1 (0.025 ± 0.008%). However, at week 4, the number of PCNA+ cardiomyocytes in the remote myocardium was about three-fold higher compared with HO-1 induced hearts (0.071 ± 0.01%, P = 0.001) (Figure 3b).
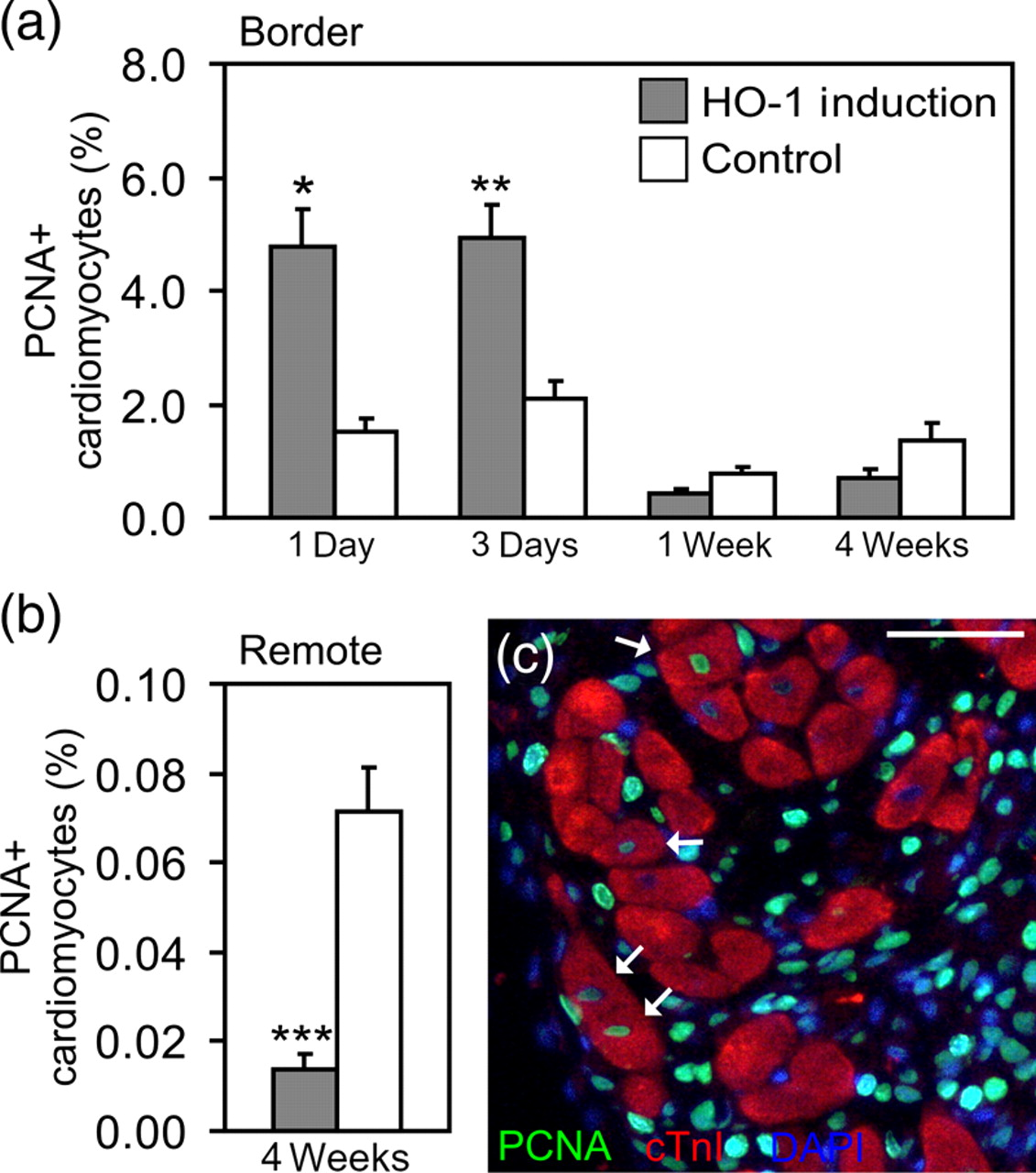
Synthesis and repair of cardiomyocyte DNA after myocardial infarction. (a) Proportion of PCNA+ cardiomyocytes in the infarct border area and (b) in the remote myocardium. Values are mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001 versus control group. Number of hearts/group is shown in Table 1. (c) PCNA+ cardiomyocytes in the infarct border area (arrows). Scale bar = 50 μm. cTnI,cardiac troponin I; PCNA, proliferating cell nuclear antigen
Effect of HO-1 induction on number and proliferation of total cells and fibroblasts
Immunohistochemical analysis of Ki67 and DAPI was performed to assess the number of all proliferating cells in comparison with the total cell count. In sham-operated hearts, the total cell number was stable throughout the study (2648 ± 81 cells/mm2) and no differences were found between the groups. In the infarcted hearts, HO-1 induction reduced the total cell number in the infarct area compared with control infarct hearts at day 3 (P = 0.007; Figure 4a), concomitantly reducing the number of proliferating cells (P = 0.025; Figure 4b). Reduced proliferation of cells in response to HO-1 induction was also observed in the infarct border area at week 1 (P = 0.004; Figure 5a) and a trend for reduced proliferation in the infarct area at week 1 (P = 0.068; Figure 4b). The complete data on cell number and proliferation of cells in infarcted hearts are shown in Supplemental Table 2 (please see Proliferation of total cells and fibroblasts in the infarct area. (a) Total number of cells and (b) proportion of Ki67+ cells in the infarct area. (c) The number of fibroblasts and (d) proportion of Ki67+ fibroblasts in the infarct area. Values are mean ± SEM. *P < 0.05 and **P < 0.01 versus control group. Number of hearts/group is shown in Table 1. (e–h) Representative images of Ki67+ fibroblasts in the infarct area at different time points. Scale bar = 50 μm Proliferation of total cells and fibroblasts in the infarct border area. (a) Proportion of Ki67+ cells in the border area. (b) The number of fibroblasts and (c) proportion of Ki67+ fibroblasts in the border area. Values are mean ± SEM. *P < 0.05 and **P < 0.01 versus control group. Number of hearts/group is shown in Table 1. (d, e) Representative images of Ki67+ fibroblasts in the infarct border area. Scale bar = 50 μm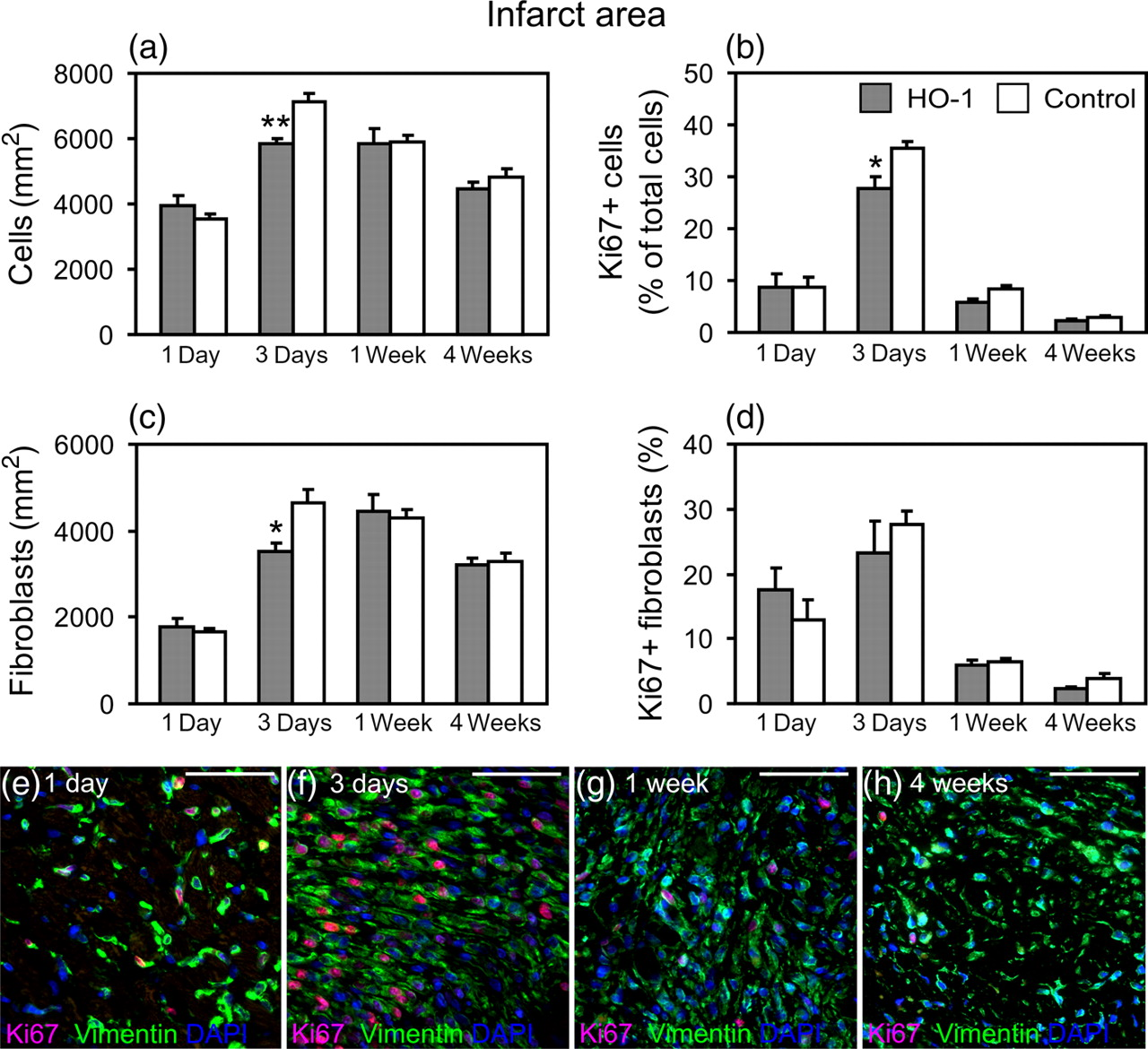
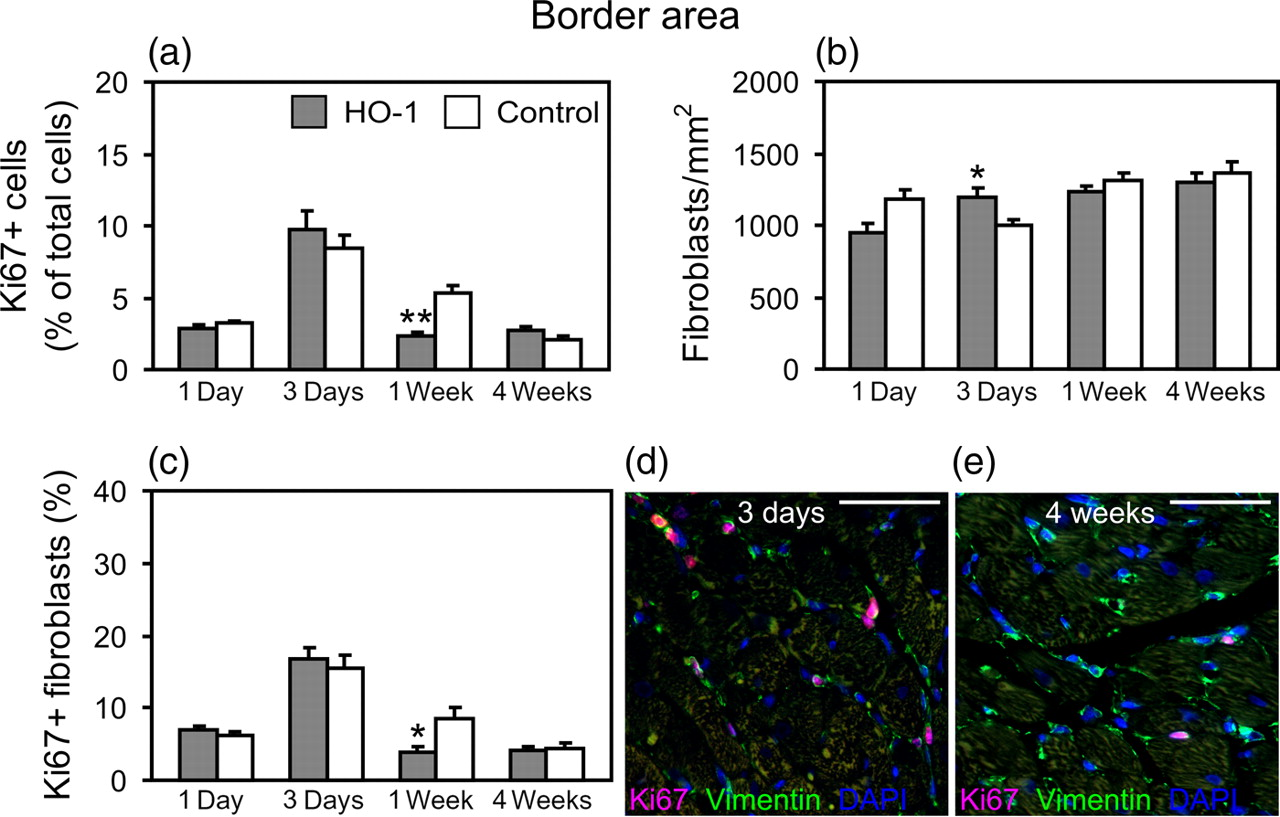
Immunohistochemical analysis of Ki67 and vimentin was performed to assess the number and proliferation of fibroblasts. The complete data on fibroblast number and proliferation are shown in Supplemental Table 3 (please see
Effect of HO-1 induction on interstitial and perivascular fibrosis and cardiomyocyte cross-sectional area
The extent of interstitial and perivascular fibrosis was assessed from the infarcted and sham-operated hearts at week 4 by picrosirius red staining. HO-1 induction decreased the ratio of perivascular fibrosis in both infarcted hearts (1.29 ± 0.06 versus 1.74 ± 0.13, P = 0.006) and sham-operated hearts at week 4 (1.14 ± 0.05 versus 1.57 ± 0.08, P = 0.002) (Figure 6b). However, there were no significant differences between the groups in interstitial fibrosis (Figure 6a).
Interstitial and perivascular fibrosis, and cardiomyocyte size. (a) Quantification of interstitial fibrosis in sham-operated and infarcted hearts at week 4 with representative images of interstitial collagen in HO-1-induced and control myocardial infarction (MI) hearts. (b) Quantification of perivascular fibrosis in sham-operated and infarcted hearts at week 4 with representative images of perivascular fibrosis in HO-1-induced and control MI hearts. (c) Cardiomyocyte cross-sectional area in HO-1-induced and control MI hearts at weeks 1 and 4, and representative images of cardiomyocytes at week 4 in HO-1-induced and control MI hearts. Cardiomyocytes were stained with anticardiac myosin (green). Values are mean ± SEM. **P < 0.01 versus control group. †
P < 0.05 and ††
P < 0.01 versus corresponding group at week 1. Number of hearts/group is shown in Table 1. Scale bar = 50 μm. HO-1, heme oxygenase-1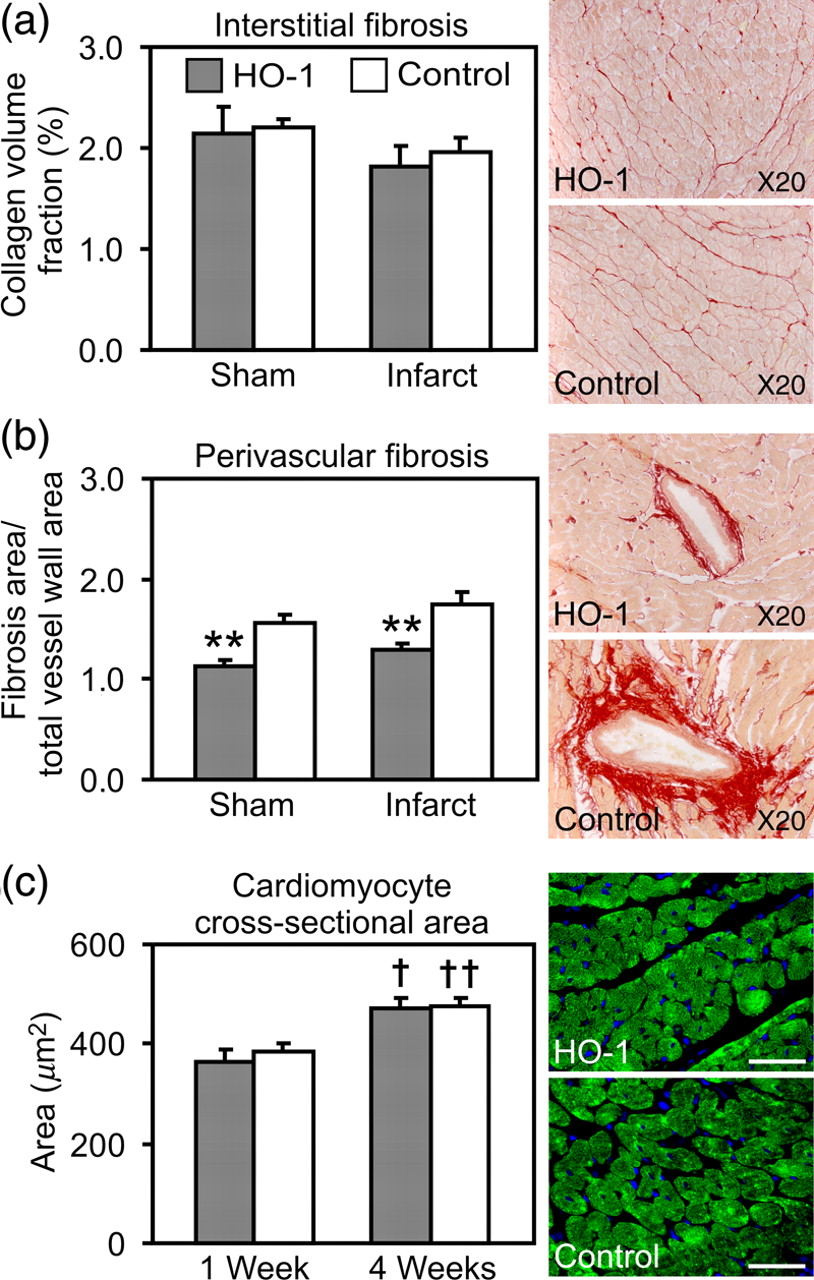
Immunohistochemical staining of cardiac myosin was performed to measure the cross-sectional area of cardiomyocytes. Cardiomyocyte cross-sectional area was significantly increased at week 4 in both HO-1-induced and control MI hearts compared with corresponding groups at week 1 (HO-1 471 ± 20 versus 364 ± 25 μm 2 , P < 0.05, and control 476 ± 15 versus 383 ± 20 μm 2 , P < 0.01; Figure 6c). However, cardiomyocyte cross-sectional area did not differ significantly between groups.
Effect of HO-1 induction on collagen, fibronectin and vimentin expression
The gene expression of major extracellular matrix proteins, procollagen type I and type III, and fibronectin was analyzed by realtime RT-PCR. HO-1 induction decreased Coll1a1 mRNA expression in the infarct area at week 1 (224 ± 34 versus 362 ± 26 × 104 mRNA copies, P = 0.017; Figure 7a). A strong trend for decreased Coll1a1 mRNA expression was also seen in the remote area at week 1 (9.0 ± 1.9 versus 31.4 ± 9.7 × 104 mRNA copies, P = 0.053; Figure 7b). There were no significant differences between the groups in the expression of procollagen type III or fibronectin mRNAs (data not shown).
Collagen and vimentin expression in the infarcted hearts. (a) Procollagen type I alpha1 mRNA expression in the infarct area, and (b) in the remote myocardium. Number of hearts/group is shown in Table 1. (c) Procollagen type I protein expression in the infarct area, and (d) in the remote myocardium with representative immunoblots. (e) Vimentin protein levels in the infarct area, and (f) in the remote myocardium with representative immunoblots. n = 6 per group in procollagen type I and vimentin protein expression analyses. Values are mean ± SEM. *P < 0.05 versus control group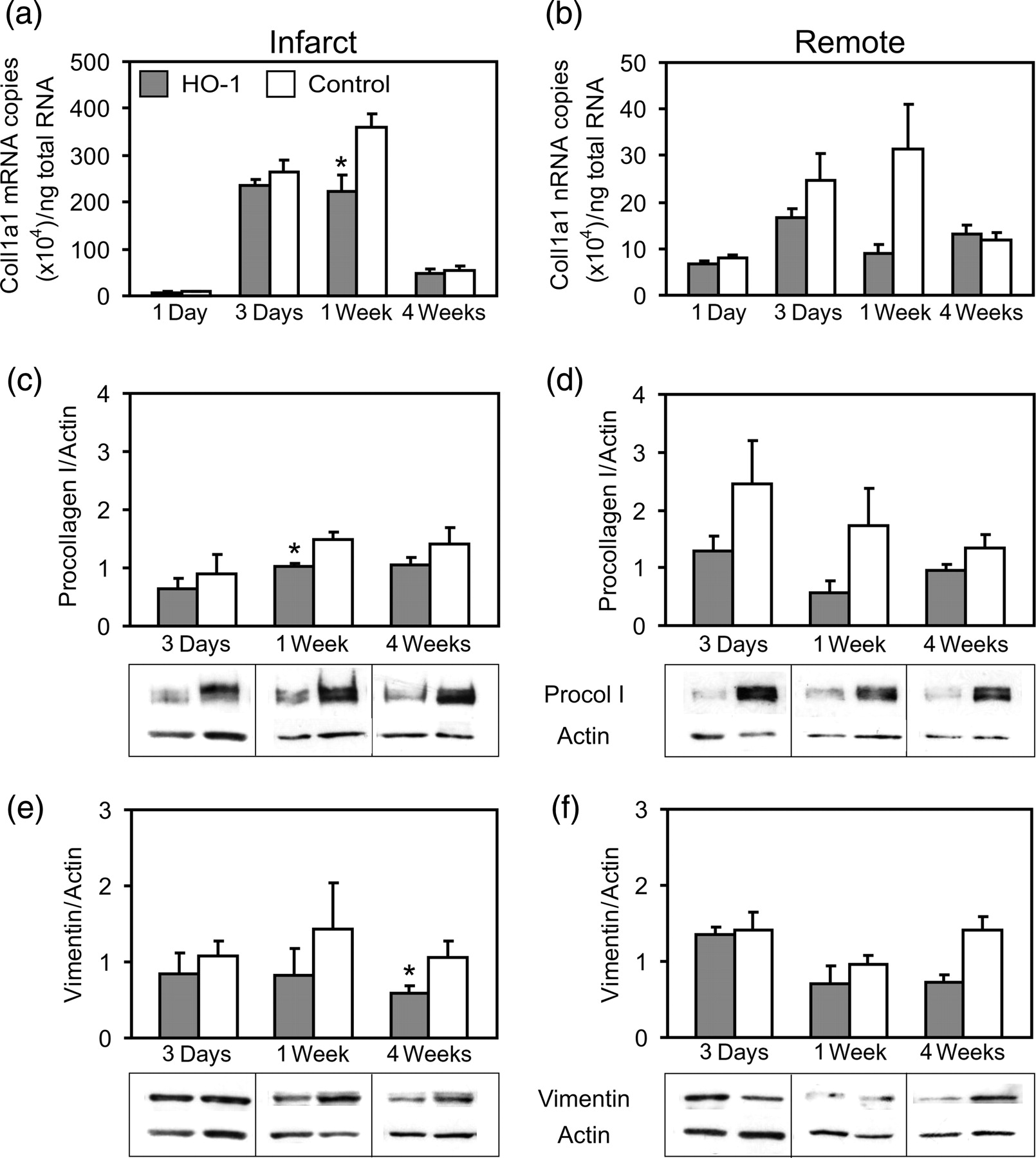
The levels of procollagen type I and vimentin proteins were analyzed by Western blotting. HO-1 induction decreased the levels of procollagen type I protein in the infarct area at week 1 (1.03 ± 0.05 versus 1.49 ± 0.13, P = 0.029; Figure 7c). There were no significant differences in the expression of procollagen type I protein in the remote area (Figure 7d). Vimentin protein levels were significantly decreased by HO-1 induction in the infarct area at week 4 (0.72 ± 0.10 versus 1.42 ± 0.17, P = 0.012; Figure 7e), and a trend for decreased vimentin expression was found in the remote area at week 4 (0.58 ± 0.11 versus 1.05 ±0.22, P = 0.093; Figure 7f).
Effect of HO-1 induction on CTGF, TGF-β1 and ANP mRNA expression
The gene expression of profibrotic factors CTGF and TGF-β1, and heart failure marker ANP were analyzed by realtime RT-PCR. HO-1 induction decreased the expression of CTGF mRNA both in the infarct area (4.0 ± 0.5 versus 6.1 ± 0.5 × 104 mRNA copies, P = 0.021; Figure 8a) and in the remote area at week 4 (2.5 ± 0.4 versus 3.8 ± 0.5 × 104 mRNA copies, P = 0.022; Figure 8b). A trend for decreased CTGF expression was also seen in the infarct area at day 1 (6.9 ± 1.1 versus 10.2 ± 0.9 × 104 mRNA copies, P = 0.068; Figure 8a). In contrast, HO-1 induction increased CTGF mRNA expression at day 3 in the remote area (3.0 ± 0.3 versus 2.0 ± 0.2 × 104 mRNA copies, P = 0.024; Figure 8b). There were no significant differences in the expression of CTGF mRNA in the infarct border area or sham-operated hearts.
CTGF, TGF-β1 and ANP mRNA expression in the infarcted hearts. (a) CTGF mRNA expression in the infarct area and (b) in the remote myocardium. (c) TGF-β1 mRNA expression in the infarct area. (d) ANP mRNA expression in the infarct border area. Values are mean ± SEM. *P < 0.05 versus control group. Number of hearts/group is shown in Table 1. CTGF, connective tissue growth factor; TGF-β1, transforming growth factor-β1; ANP, atrial natriuretic peptide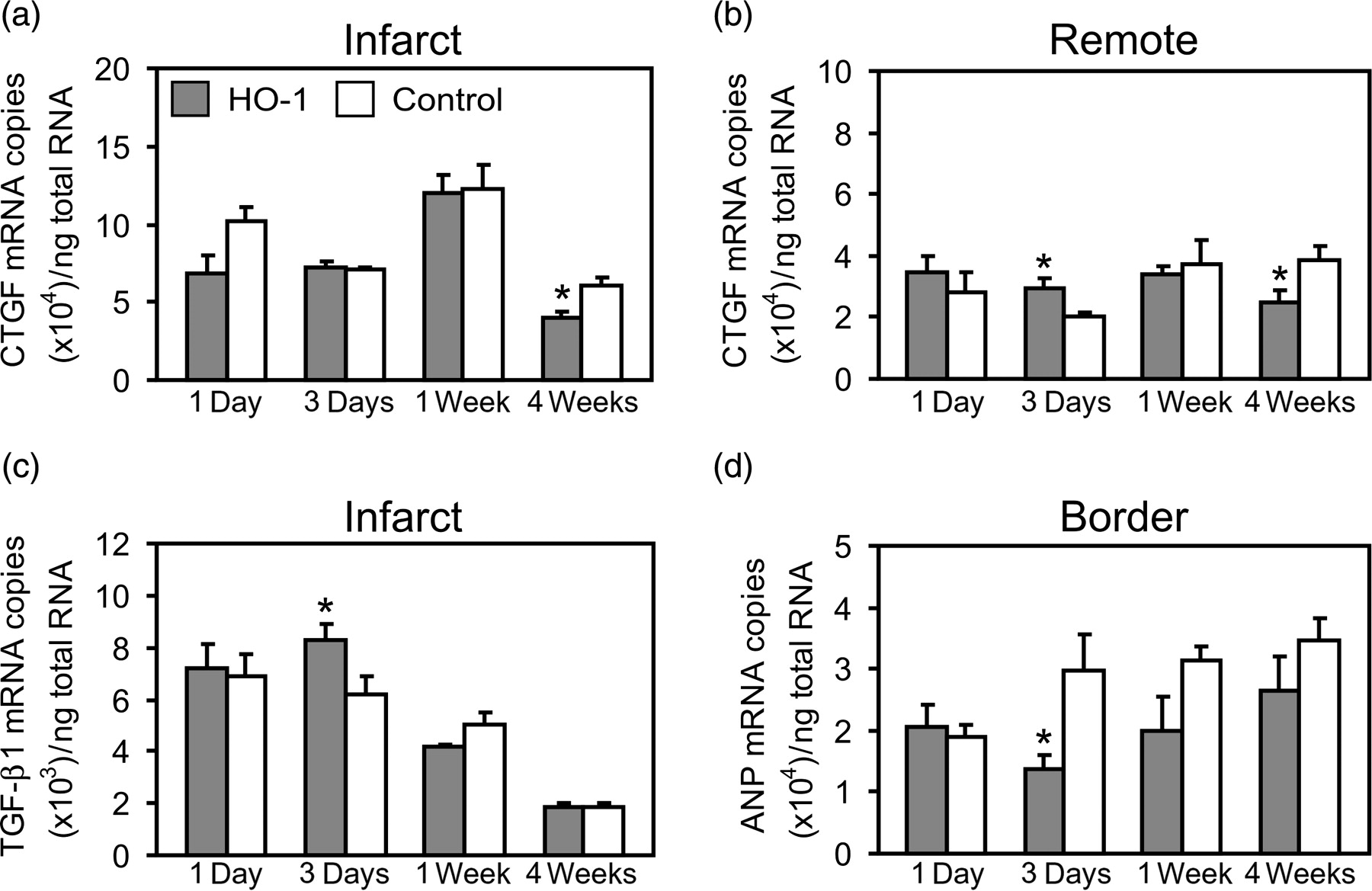
HO-1 induction increased TGF-β1 mRNA expression in the infarct area at day 3 (8.3 ± 0.6 versus 6.2 ± 0.7 × 103 mRNA copies, P = 0.037; Figure 8c). There were no significant differences in the expression of TGF-β1 mRNA in the infarct border or remote areas or sham-operated hearts.
The expression of ANP mRNA was decreased in the infarct border area at day 3 (1.4 ± 0.2 versus 3.0 ± 0.6 × 104 mRNA copies, P = 0.046) and week 1 (2.0 ± 0.5 versus 3.1 ± 0.3 × 104 mRNA copies, P = 0.050) in HO-1 induced hearts (Figure 8d). The expression of ANP mRNA did not differ significantly in the infarct or remote areas, or in the sham-operated hearts.
Discussion
The present study demonstrates that HO-1 has an important role in maintaining cellular homeostasis in the postischemic heart. Furthermore, the overall effect of HO-1 appears to depend on the prevailing milieu of cells and factors secreted by them in the infarcted heart. We found that HO-1 induction acted cardioprotectively by increasing proliferation and repair of cardiomyocytes, and decreasing the apoptotic loss of cardiomyocytes in the infarct border area during the first few days after MI. Concomitantly, HO-1 decreased proliferation of fibroblasts and expression of procollagen type I and increased expression of TGF-β1 in the infarct area, suggesting that the beneficial effect of HO-1 in the early phase of infarct healing may result partly from the suppression of the inflammatory response via TGF-β1. In the remote myocardium, HO-1 induction inhibited both proliferation and apoptosis of cardiomyocytes and attenuated the heart failure-induced increase in the repair of cardiomyocyte DNA, thereby pointing to a protective role for HO-1 in the remote myocardium as well. In addition, HO-1 induction inhibited the proliferation of fibroblasts, modulated the expression of extracellular matrix components and reduced perivascular fibrosis during the healing of infarcted myocardium, possibly via down-regulation of profibrotic growth factor CTGF.
This is the first study to show the temporal and spatial effects of HO-1 induction on proliferation of cardiomyocytes in the postinfarction hearts. Previous in vitro studies have shown that HO-1 increases proliferation of endothelial cells whereas it decreases proliferation of several other cell types, including vascular and airway smooth muscle cells, cardiac and lung fibroblasts and pancreatic stellate cells. 13,14,16,21–23 We have recently shown that pretreatment with the HO-reaction product CO increased the number of proliferating cardiomyocytes concomitantly with the increase in c-kit+ stem/progenitor cells in the infarcted rat hearts. 8 However, the increased proliferation of cardiomyocytes in CO-donor pretreated hearts represented proliferation of newly formed cardiomyocytes derived from c-kit+ cells rather than specific regulation of the cardiomyocyte cell cycle by CO. In the present study, we show in rat hearts in vivo, that HO-1 may also potentially regulate the cardiomyocyte cell cycle. Moreover, the overall effect of HO-1 induction on the number of proliferating cardiomyocytes appears to be dependent on the milieu of cardiomyocytes and soluble factors at a given time and location in the postischemic heart.
It has been shown previously that the heart contains a population of proliferative cardiomyocytes, and the proliferation of cardiomyocytes is increased in response to myocardial infarction. 24–26 We found that HO-1 induction significantly increased the number of Ki67+ proliferating cardiomyocytes in the infarct border area during the first few days after MI. This suggests that HO-1 may protect the cardiomyocytes and maintain cellular homeostasis in the ischemic conditions, thereby increasing proliferation of cardiomyocytes, because unlike in the infarct border area, HO-1 induction inhibited proliferation of cardiomyocytes in the supposedly normal conditions in remote myocardium. The cardioprotective effect of HO-1 in the early phase of infarct healing could be mediated by the antioxidative action of biliverdin and bilirubin. Bilirubin has earlier been shown to decrease infarct size after I/R in rats. 27 In contrast, the significantly lower number of proliferating cardiomyocytes in the remote myocardium of HO-1-induced hearts suggests indirectly that HO-1 may inhibit the progression of the cardiomyocyte cell cycle in normal conditions. This hypothesis is supported by earlier in vitro studies showing that HO-1 inhibits proliferation of other muscle cell types, VSMCs and airway SMCs. 21,22 However, further studies in cardiomyocytes in vitro are needed to determine the effect of HO-1 on cardiomyocyte proliferation in different conditions. Nevertheless, these opposing spatiotemporal effects of HO-1 on cardiomyocyte proliferation are likely to result in the enhanced recovery of myocardial infarction.
The cardioprotective action of HO-1 induction is supported by the coincident increase in PCNA+ cardiomyocytes in the infarct border area. As Ki67 is a marker of DNA synthesis, PCNA is a marker of both DNA synthesis and repair. 28,29 The number of PCNA+ cardiomyocytes was three-fold higher than the number of Ki67+ cardiomyocytes in the sham-operated hearts and 5–10-fold higher in the infarcted hearts, indicating that a major part of PCNA+ cardiomyocytes were under DNA repair while only a small part of PCNA+ cardiomyocytes were proliferating cells. In addition, the repair of cardiomyocyte DNA was increased in the infarcted hearts, particularly in the infarct border areas compared with sham-operated hearts. Furthermore, we show for the first time, that HO-1 induction significantly enhanced the repair of cardiomyocyte DNA in the infarct border areas during the first few days after MI. This finding further supports the role for HO-1 in maintaining cellular homeostasis in the early phase of infarct healing. The increase in PCNA+ cardiomyocytes may also serve as a marker of stress and the development of heart failure. Increased repair of cardiomyocyte DNA has earlier been reported in hearts with dilated cardiomyopathy and severe heart failure, 30,31 whereas implantation of a left ventricular assist device has been shown to reduce PCNA expression. 31 In our study, HO-1 induction attenuated the increase in PCNA+ cardiomyocytes at week 4 in the remote myocardium, suggesting that HO-1 may protect against the adverse ventricular remodelling and development of heart failure in the late phase of infarct healing, if increased PCNA expression is considered as a marker of heart failure.
The third facet in the cardioprotection observed in our study is the well known antiapoptotic property of HO-1. HO-1 gene transfer and HO-1 induction by CoPPIX have been shown to inhibit apoptosis of cardiomyocytes. 32,33 HO-1 induction promoted survival of cardiomyocytes by decreasing cardiomyocyte apoptosis in our study. The reduced cardiomyocyte loss, together with increased proliferation and repair of existing cardiomyocytes, further strengthens the important role for HO-1 in maintaining homeostasis of cardiomyocytes in the area at risk. Thus, HO-1 may prevent the expansion of the infarct area and even favor survival and regeneration of myocardium over cardiomyocyte loss as suggested by the trend of smaller infarcts in the HO-1-induced hearts at weeks 1 and 4 after MI. Furthermore, the antiapoptotic action of HO-1 in the remote myocardium potentially attenuates the progression of adverse ventricular remodelling.
In addition to cardiomyocytes, cardiac fibroblasts are an equally important cell type in the heart. Cardiac fibroblasts are the most abundant cell type in the rat heart, 34 and they have a central role in the post-MI ventricular remodelling. 35 Fibroblasts migrate into the infarct area, proliferate and participate in the repair of injury by forming a fibrous scar. 35,36 However, inappropriate activation of fibroblasts and increased fibrosis together with apoptosis and hypertrophy of cardiomyocytes leads to the development of heart failure. 35,36 We found a lower number of both total cells and vimentin+ fibroblast-like cells in the infarct areas of HO-1-induced hearts at day 3, and lower levels of vimentin protein (an indirect measure of fibroblast number) at week 4, suggesting that HO-1 may inhibit proliferation of cardiac fibroblasts. HO-1 gene transfer has previously been shown to inhibit proliferation of cardiac fibroblasts in vitro, and decrease the infiltration of myofibroblasts in the infarct area in vivo. 14 Likewise, we found that HO-1 induction decreased the proportion of both proliferating total cells and proliferating fibroblasts in the infarct area and infarct border area. Furthermore, HO-1 induction decreased the expression of procollagen type I mRNA and protein at week 1, and the extent of perivascular fibrosis at week 4. The antifibrotic effect of HO-1 has earlier been demonstrated in the kidneys, liver and lungs, 37–39 and HO-1 induction by hemin has been shown to attenuate left ventricular hypertrophy and fibrosis in adult spontaneously hypertensive rats. 11 In addition, HO-1 gene transfer has been shown to decrease interstitial collagen at 1.5 and three months after I/R injury in rat hearts. 14 Although HO-1 decreased perivascular fibrosis in our study, it had no effect on the extent of interstitial fibrosis or on the expression of collagen type III or fibronectin mRNAs that are usually increased in interstitial fibrosis. We also found no differences in cardiomyocyte cross-sectional area. However, these controversies may be due to the different models or more likely, the different time points studied, as week 4 may be too early to see differences in interstitial fibrosis or in cardiomyocyte hypertrophy. Interstitial fibrosis is also more characteristic of hypertension and diabetes than MI alone. Therefore, it is possible that interstitial fibrosis will not always increase in previously healthy Wistar rats, as in this study. This is supported by our earlier study showing that interstitial fibrosis was not increased in vehicle-treated Wistar rats even at 12 weeks' post-MI compared with sham-operated, vehicle-treated hearts. 40 In addition, there is earlier evidence that HO-1 does not affect cardiomyocyte hypertrophy. 9
TGF-β is a key factor regulating both the healing of MI and post-MI ventricular remodelling. 41 Although TGF-β is known to promote cardiomyocyte hypertrophy and increase fibrosis in the late phases of infarct healing, it has a crucial anti-inflammatory role in the early phase of infarct healing. 41 TGF-β suppresses the inflammatory response and may also participate in limiting the expansion of inflammation into the non-infarcted myocardium. 41,42 We found that HO-1 induction increased the gene expression of TGF-β1 in the infarct area and CTGF in the remote area at day 3. These results corroborate the anti-inflammatory role of HO-1 in the early phase of infarct healing and suggest that HO-1 may partly maintain cellular homeostasis in the infarcted heart by modulating the expression of anti-inflammatory cytokine TGF-β1. Furthermore, the simultaneous increase in CTGF expression (one of the downstream mediators of the profibrotic effect of TGF-β) may relate to the progression of infarct healing from the inflammatory phase to the proliferative phase characterized by suppression of the inflammatory response and formation of the fibrous scar. TGF-β and IL10 have earlier been shown to mediate the anti-inflammatory action of HO-1 against ovalbumin-induced airway inflammation. 43 In contrast, HO-1 induction by heme arginate has been shown to decrease cardiac TGF-β levels and fibrosis in deoxycorticosterone acetate-salt hypertension. 44 In our study, HO-1 had no effect on TGF-β1 expression at later time points. Instead, we found that HO-1 significantly decreased gene expression of CTGF at week 4 in both the infarct area and in the remote myocardium, suggesting that in the late phases of infarct healing, HO-1 may protect against fibrosis by down-regulating CTGF. To our knowledge, this is the first study to show that HO-1 potentially regulates the expression of CTGF.
Finally, we investigated the expression of ANP, a marker of increased cardiac load and heart failure. ANP is increased in response to MI, and pharmacological administration of ANP in an ischemia–reperfusion setting has been shown to reduce infarct size and inhibit cardiomyocyte hypertrophy and fibrosis. 45 In contrast, ANP has also been shown to have proinflammatory effects, 46 and ANP was recently shown to increase neutrophil infiltration, infarct size and mortality after LAD ligation. 47 ANP has earlier been shown to induce HO-1 in the liver and in endothelial cells, 48,49 whereas HO-1 was recently shown to decrease ANP expression in pressure-overloaded mice hearts. 50 Likewise, in our study, HO-1 induction attenuated the MI-induced increase in ANP gene expression in the infarct border area. Whether HO-1 specifically down-regulates the expression of ANP, and thereby abolishes the proinflammatory effect of ANP, or whether the attenuated ANP expression simply reflects the cardioprotective effect of HO-1 remains to be determined in further studies.
This study has some limitations. One limitation is the lack of echocardiography measurements. Despite the several beneficial effects of HO-1 in post-MI hearts, we found no significant effect on infarct size. A trend for smaller infarcts in HO-1-induced hearts suggests that HO-1 may have prevented the expansion of ischemic injury by promoting survival and proliferation of cardiomyocytes in the infarct border area. Interestingly, Wang et al. 51 showed recently that the infarct size was similar in cardiac-restricted HO-1-transgenic mice compared with non-transgenic mice, although HO-1 overexpression alleviated ventricular remodelling and improved cardiac function four weeks after permanent LAD ligation. Therefore, it appears that the salutary effects of HO-1 and CO do not always result in decreased infarct size in the permanent LAD ligation model. In addition, week 4 may be too early to detect improvement in infarct size. A second limitation is that we did not use HO enzyme inhibitors to abolish the beneficial effects of HO-1. This needs to be done in further studies, as well as using the new CO releasing molecules and biliverdin to investigate the molecular mechanisms of HO-1. A third limitation is that HO-1 may have modulated cardiac remodelling by regulating the levels of heme and thereby the levels of heme-dependent proteins, such as nitric oxide synthase, cyclooxygenase and NADPH oxidase. HO-1 has earlier been shown to modulate the levels of these proteins. 52–55 The roles of heme-dependent proteins in HO-1-induced cardioprotection remains to be investigated in further studies.
In conclusion, HO-1 induction protected the heart at a number of levels during the healing of myocardial infarction. It modulated the survival, proliferation and repair of cardiomyocytes, and decreased fibrosis and proliferation of fibroblasts in a manner dependent on the prevailing milieu of cells in the infarcted heart. Our results show that the induction of HO-1 is potentially useful for enhancing the recovery of myocardial infarction and protecting against pathological changes in the myocardium.
Footnotes
ACKNOWLEDGEMENTS
We thank Riina Hatakka, Katariina Immonen, Riikka Kosonen, Jarkko Lakkisto and Terhi Suvanto for expert technical assistance. This work was supported in part by grants from the Helsinki University Central Hospital, the Finnish Foundation for Cardiovascular Research, the Sigrid Jusélius Foundation, the Koskelo Foundation, the Aarne and Aili Turunen Foundation, the Finnish Foundation for Medical Research, the Finnish-Norwegian Medical Foundation, the Finnish Foundation for Laboratory Medicine and the National Graduate School of Clinical Investigation.