
Abstract
Obesity is a major risk factor for coronary artery disease, but its impact on anesthetic-induced cardioprotective actions is unexplored. We tested whether obesity inhibits anesthetic sevoflurane-induced preconditioning and whether this effect is mediated via the AMP-activated protein kinase (AMPK) signaling pathway. Sprague-Dawley rats were fed a high-fat (HF, 45% kcal as fat) or low-fat (LF, 10% kcal as fat) diet for 12 weeks. HF-fed rats developed metabolic disturbances including visceral obesity, hyperinsulinemia, hyperleptinemia and dyslipidemia. HF- or LF-fed rats subjected to 25 min of myocardial ischemia followed by 120 min of reperfusion were assigned to the following groups: control, sevoflurane preconditioning, sevoflurane plus AMPK inhibitor ara-A or AMPK activator A769662 alone. Infarct size was similar between the two control groups. Sevoflurane preconditioning significantly reduced infarct size in LF-fed rats but failed to induce cardioprotection in HF-fed rats. Phosphorylation of AMPK and endothelial nitric oxide synthase, as well as myocardial nitrite and nitrate, were also increased by sevoflurane preconditioning in LF-fed rats but not in HF-fed rats. Pretreatment with ara-A inhibited phosphorylation of AMPK and reversed sevoflurane preconditioning-induced cardioprotection in LF-fed rats, whereas it had no effects in HF-fed rats. In addition, sevoflurane preconditioning failed to enhance reactive oxygen species (ROS) generation in the myocardium of HF-fed rats compared with LF-fed rats. Direct activation of AMPK with A769662 equally increased phosphorylation of AMPK and reduced infarct size in both LF- and HF-fed rats. The results suggest that diet-induced obesity suppresses sevoflurane preconditioning-induced cardioprotective action, probably due to a diminished effect of sevoflurane preconditioning on activation of the ROS-mediated AMPK signaling pathway.
Keywords
Introduction
High dietary intake of fats may lead to several serious metabolic defects, including obesity, which is a major health issue in Western countries. This pathology constitutes an important risk factor for coronary artery disease and is associated with increased cardiovascular mortality and worse clinical outcomes following myocardial infarction. 1 Therefore, reducing the consequences of coronary artery disease using any effective cardioprotective strategy should be investigated in this population. A rapidly growing body of evidence indicates that volatile anesthetics produce pharmacological preconditioning and protect the heart against myocardial infarction in a variety of experimental animal models as well as in humans. 2–7 Although multiple signaling pathways have been suggested to mediate anesthetic-induced preconditioning, the exact mechanism is not yet fully understood. Anesthetic-induced preconditioning may be more clinically relevant than ischemic preconditioning because it avoids the vascular and myocardial injury that could result from coronary artery occlusion, and therefore are less harmful. Unfortunately, there is no study that has addressed the issue of anesthetic preconditioning in obesity.
AMP-activated protein kinase (AMPK) is an important sensor of cellular energy status and has a key role in regulating cellular energy metabolism. 8,9 In response to a fall in intracellular ATP levels, it activates energy-producing pathways and inhibits energy-consuming processes. 8,9 AMPK has been implicated in a number of diseases related to energy metabolism, including obesity. Recent evidence indicates that AMPK is involved in anesthetic-induced cardioprotection and this activation requires upstream production of reactive oxygen species (ROS). 10,11 Activation of AMPK may lead to increased levels of endothelial nitric oxide synthase (eNOS) and stimulate nitric oxide (NO) production, 12,13 which is an important trigger and mediator of endogenous cardioprotective signal transduction in both ischemic and anesthetic-induced preconditioning. 4,14,15 It is unknown whether the AMPK signaling pathway mediates anesthetic preconditioning-induced cardioprotective effects in obesity, where intrinsic AMPK activation has been found to be depressed. 16
Therefore, the aims of our present study were to determine whether diet-induced obesity influences volatile anesthetic sevoflurane-induced cardioprotective actions against ischemia–reperfusion injury in vivo and to evaluate whether this effect is mediated via the AMPK signaling pathway. For this purpose, we chose the high-fat diet-induced obese rat model, which mimics human obesity syndrome. 17
Methods
Animals
Weanling male Sprague-Dawley rats weighing 85.6 ± 0.7 g (Beijing Laboratory Animal Research Center, Beijing, China) were randomly assigned to receive a high-fat diet (HF; 45% kcal as fat; Research Diets, New Brunswick, NJ, USA) or a low-fat diet (LF; 10% kcal as fat) for 12 weeks. All rats were housed in a room maintained at 23–25°C with a 12-h light/dark cycle and were given food and water ad libitum. These studies were performed in accordance with the ‘Guiding Principles for Research Involving Animals and Human Beings’. 18 The experimental procedures were approved by the Institutional Animal Care and Use Committee of the China Medical University.
Surgical preparation
Rats were anesthetized (ketamine 90 mg/kg + xylazine 10 mg/kg intraperitoneally) and were mechanically ventilated with oxygen-enriched air after tracheal intubation. The respiratory rate was adjusted to maintain partial pressure of carbon dioxide within physiological limits. A thermistor probe was inserted 8.0 cm into the rectum to monitor the core body temperature that was maintained at normothermia between 37.0 and 38.0°C with a circulating warm water (37.5°C) underbody heating pad. After a midline cervical incision, the right jugular vein and carotid artery were cannulated for fluid replacement and drug administration or for measurement of aortic pressure, respectively. Aortic pressure signals were digitized using an analog–digital converter and continuously recorded on a computer using the PowerLab software (PowerLab/8SP, Chart 5.0; ADInstruments Pty, Ltd, Castle Hill, Australia). Anesthesia was maintained by continuous [alpha]-chloralose infusion (25 mg kg BW−1 h−1). After left-sided lateral thoracotomy and pericardiotomy were performed, a 6-0 silk suture was passed below a main branch of the left coronary artery. The ends of the suture were passed through a propylene tube to form a snare. Successful coronary occlusion was verified by epicardial cyanosis.
Experimental protocol
The experimental design is illustrated in Figure 1. Rats were randomly assigned to eight groups, and after a 30-min stabilization period, all rats were subjected to 25 min of regional myocardial ischemia followed by 120 min of reperfusion. At the end of the protocol, the blood samples were collected from the right carotid artery for biochemical measurements; the heart and epididymal fat were removed and weighed, and the hearts were then processed for measurements of area at risk and infarct size.
Schematic diagram illustrating the experimental protocol. Animals were subjected to 25 min of regional myocardial ischemia and 120 min of reperfusion. Three five-minute periods of one minimum alveolar concentration sevoflurane, interspersing with two five-minute washout periods, were performed 10 min before coronary occlusion. AMPK (AMP-activated protein kinase) inhibitor ara-A (100 μg/kg/min, intravenously) was administered for 10 min before sevoflurane preconditioning; AMPK activator A-769662 (6 mg/kg, intraperitoneally) was administered 30 min before ischemia. LF, low-fat-fed rats; HF, high-fat-fed rats; Sevo, sevoflurane; A769, A769662. (A color version of this figure is available in the online journal)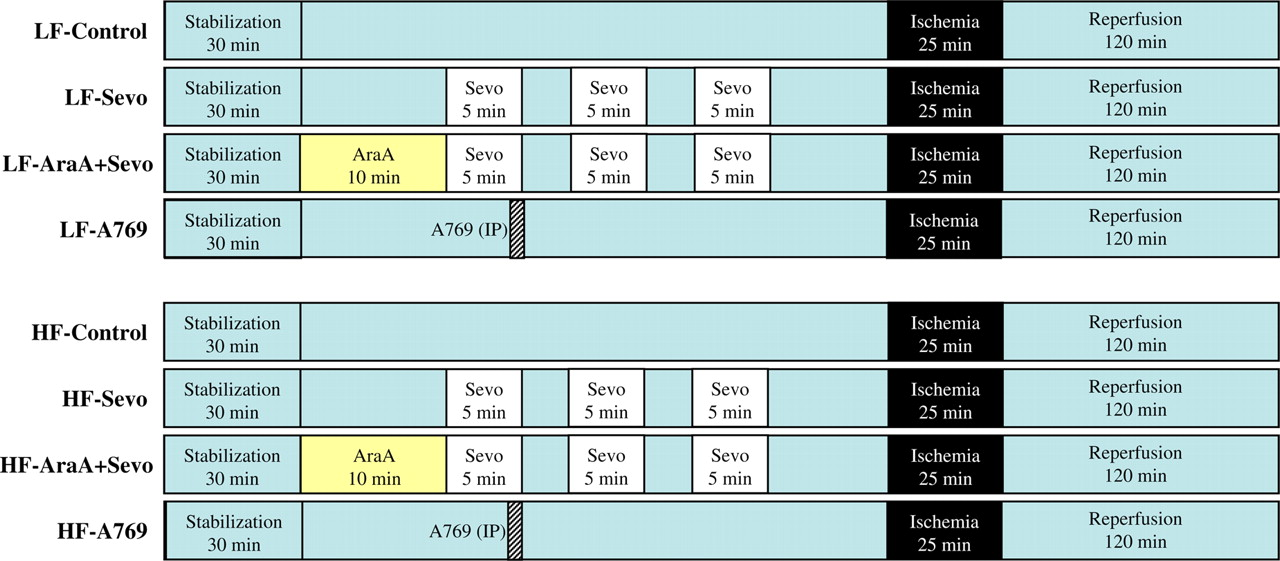
The eight groups were:
LF-fed ischemia–reperfusion alone group (LF-Control, n = 10); LF-fed sevoflurane preconditioning group (LF-Sevo, n = 10). Rats received 1 minimum alveolar concentration sevoflurane (in rats 2.4 vol%) for three five-minute periods, interspersed with two five-minute washout periods, 10 min before a 25-min duration of left coronary artery occlusion. The dose for sevoflurane was chosen according to a previous study in which this dose was determined to result in optimal cardioprotective actions in vivo in rats;
19
LF-fed sevoflurane preconditioning plus ara-A group (LF-AraA + Sevo, n = 10). Same as group (2), but treated with ara-A, an inhibitor of AMPK (100 μg/kg/min, intravenously) for 10 min before preconditioning;
12
LF-fed A-769662 alone group (LF-A769662, n = 8). Rats were treated with A-769662, an activator of AMPK (6 mg/kg, intraperitoneally), 30 min before a 25-min duration of left coronary artery occlusion;
20
HF-fed ischemia–reperfusion alone group (HF-Control, n = 10); HF-fed sevoflurane preconditioning group (HF-Sevo, n = 10); HF-fed sevoflurane preconditioning plus ara-A group (HF-AraA + Sevo, n = 10); HF-fed A-769662 alone group (HF-A769662, n = 8).
To investigate the role of the AMPK and NO signaling pathways in anesthetic preconditioning, additional experiments were performed. Rats underwent the same protocol as described above (n = 10 per group). At the end of ischemia, rats were sacrificed, and myocardial samples were collected from ischemic left ventricular regions. Samples were then frozen in liquid nitrogen for analysis of ROS or stored at −80°C for Western blot analysis and myocardial NO measurement. Because a previous study
10
showed that the sevoflurane-induced AMPK activation occurs during ischemia, not prior to ischemia, we therefore selected this time point to collect tissues.
Measurements of infarct size and area at risk
Infarct size and area at risk were determined as described previously. 3 Briefly, at the end of the experimental protocol, hearts were excised and mounted on a Langendorff apparatus. The coronary artery was re-occluded and 0.1% methylene blue was infused into the aortic root to label the non-ischemic, blue-stained, normal area, thereby delineating the risk zone as a non-stained area. The hearts were then removed from the Langendorff apparatus, trimmed of atria and great vessels, weighed and frozen (in a cold chamber at −18°C). Hearts were then cut into 2-mm transverse slices. The slices were incubated at 37°C for 20 min in 1% 2,3,5-triphenyl tetrazolium chloride buffer. The slices were then placed in 10% formaldehyde for 10 min to increase the contrast between stained tissue as a deep red color and non-stained tissue. Slices were then photographed, and the percentages of the at-risk and infarcted areas were calculated using an image analysis program.
Western blot analysis
Myocardial samples were homogenized and proteins were extracted as described previously. 21,22 Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. Membranes were immunoblotted with antibodies anti-phosphorated (Ph)-AMPK at Thr172, anti-Ph-AMPK at Ser485/491, anti-total AMPK, anti-Ph-LKB1 (LKB1, serine/threonine kinase 11), anti-total LKB1, anti-Ph-eNOS and anti-total eNOS (Cell Signalling, Beverly, MA, USA). Protein bands were detected by enhanced chemiluminescence and analyzed by imaging densitometry.
Hemodynamic measurements and biochemical parameters
Heart rate and arterial blood pressure were digitized using an analog–digital converter and recorded on a computer using the PowerLab software (PowerLab/8SP, Chart 5.0; ADInstruments Pty, Ltd). Glucose concentrations were measured immediately after sampling with a glucose test meter (Glutest EII; Kyoto First Scientific, Kyoto, Japan). Total cholesterol and triglyceride concentrations were determined with commercially available kits (Wako Pure Chemical Industries, Osaka, Japan). Plasma levels of leptin, insulin and adiponectin were measured with commercial enzyme-linked immunosorbent assay kits (Morinaga, Kanagawa, Japan; Shibayagi, Gunma, Japan; Assaypro, St Charles, MO, USA).
Measurement of myocardial level of nitrite and nitrate (NO x )
Total level of nitrite and nitrate (NO x ) were determined in left ventricular myocardium using a nitrate/nitrite colorimetric assay kit (Cayman Chemical, Ann Arbor, MI, USA).
Dihydroethidium staining
Dihydroethidium (DHE), an oxidative fluorescent dye, was used to detect superoxide in situ in ischemic myocardium by a laser scanning confocal microscope. 23 Briefly, the heart was removed and immediately frozen at −80°C for one hour, blocked in the coronal plane and sectioned into 30-μm slices with a cryostat. The sections were mounted on the microscope slides and incubated with DHE (5 μmol/L; Molecular Probes Inc, Eugene, OR, USA) for 30 min at 37°C in a light-protected humidified chamber. Images were visualized and fluorescence was analyzed as described in a previous study by others. 23
Statistical analysis
All data are expressed as mean ± SEM. Changes in hemodynamics between groups or between time points in a group were performed using two-way analysis of variance followed by Tukey's post hoc test. Data for infarct sizes, Western blots, biochemical parameters, NO x concentrations or DHE fluorescence were analyzed by Student's t-test with Bonferroni's correction for multiple comparisons. P < 0.05 was considered statistically significant.
Results
Physiological and metabolic characterization
Characteristics of LF- and HF-fed rats at the termination of the study
LF, low-fat-fed rats; HF, high-fat-fed rats; Sevo, sevoflurane; A769, A769662
Values are mean ± SEM; n = 8–10 for each group
*P < 0.05 versus respective LF
Metabolic parameters in each group
LF, low-fat-fed rats; HF, high-fat-fed rats
Values are mean ± SEM; n = 8–10 for each group
*P < 0.05 versus respective LF
Systemic hemodynamics
Hemodynamic parameters in each group
LF, low-fat-fed rats; HF, high-fat-fed rats; HR, heart rate; mean BP, mean blood pressure; baseline, stabilization period; preischemia, five minutes before ischemia. Values are mean ± SEM; n = 8–10 for each group
*P < 0.05 versus baseline
Effect of sevoflurane preconditioning on infarct size
The area at risk and myocardial infarct size are shown in Figure 2. The ratio of area at risk did not differ (P = not significant) among groups following ischemia–reperfusion. The infarct size in LF-Control rats was similar to that in HF-Control rats. Anesthetic preconditioning with sevoflurane significantly reduced infarct size by 49% in LF-fed rats (43 ± 3% versus 22 ± 2%, P < 0.05; LF-Control versus LF-Sevo, respectively). In contrast, this strategy failed to protect HF-fed rats against myocardial infarction (45 ± 3% versus 42 ± 4%, P = not significant; HF-Control versus HF-Sevo, respectively). Administration of the AMPK inhibitor ara-A before ischemia eliminated the cardioprotection produced by sevoflurane preconditioning in LF-fed rats (45 ± 4%, P < 0.05; versus LF-Sevo), but had no effect on HF-fed rats (47 ± 5%, P = not significant; versus HF-Sevo). Administration of the AMPK activator A-769662 alone before ischemia significantly reduced infarct size in both LF-fed rats by 51% (21 ± 3%, P < 0.05; versus LF-Control) and HF-fed rats by 33% (30 ± 2%, P < 0.05; versus HF-Control), respectively.
Area at risk (a) expressed as percent of left ventricle, and infarct size (b) expressed as percent of the area at risk after 25 min of ischemia followed by 120 min of reperfusion in each group. Values were expressed as mean ± SEM (n = 8–10 for each group). *P < 0.05 versus respective control, †
P < 0.05 versus respective LF. LF, low-fat-fed rats; HF, high-fat-fed rats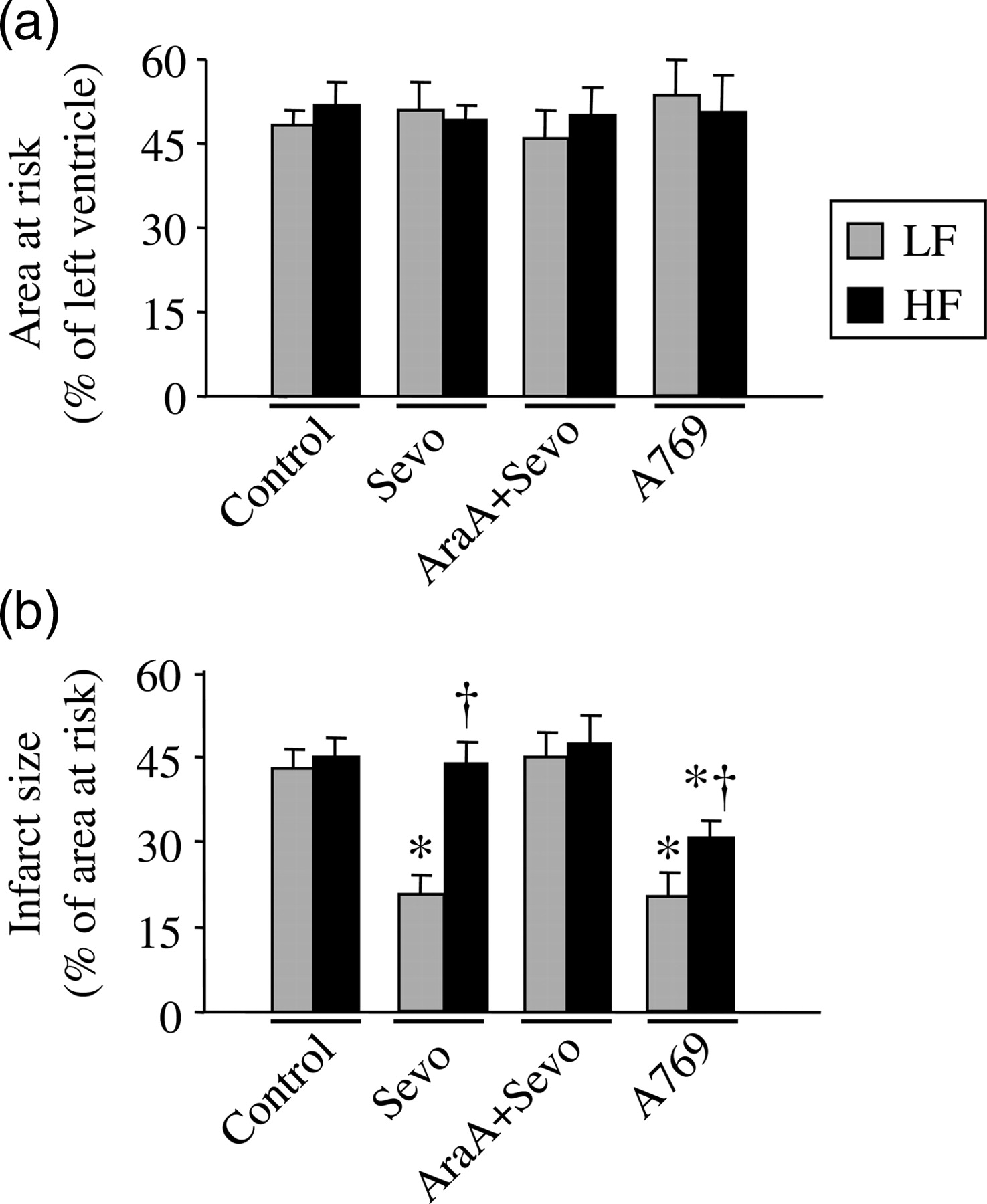
Phosphorylation of AMPK and LKB1
Phosphorylation of AMPK at Thr172 (AMPKThr172) by upstream AMPK kinase accounts for most of the activation of AMPK. 24 Phosphorylation of AMPK at Ser485/491 (AMPKSer485/491) in response to insulin in the heart decreases Thr172 phosphorylation by upstream kinase LKB1 and hence AMPK activation during ischemia. 24 We therefore evaluated phosphorylation of AMPKThr172, AMPKSer485/491 and LKB1.
As shown in Figure 3, there were no differences in Ph-AMPKThr172 (normalized to total) between the two control groups. Sevoflurane preconditioning increased (P < 0.05 versus LF-Control) Ph-AMPKThr172 in LF-fed rats, whereas it had no significant effect on HF-fed rats. Pretreatment with AMPK inhibitor ara-A reversed the sevoflurane-induced increase in Ph-AMPKThr172 in LF-fed rats but did not change that in HF-fed rats. Administration of AMPK activator A-769662 alone equally induced a robust increase in Ph-AMPKThr172 in both LH- and HF-fed rats. No significant difference in Ph-AMPKSer485/491 or Ph-LKB1 was observed among groups.
Quantitative comparison of phosphorylation (Ph-) levels of AMPKThr172 (a), AMPKSer485/491 (b) and LKB1 (c) in the ischemic myocardium from each group. Representative Western blots are aligned with the matching grouped data. Levels of phosphorylated proteins were normalized to their total protein concentrations. Values are expressed as means ± SEM (n = 6 for each group). *P < 0.05 versus respective control, †
P < 0.05 versus respective LF. LF, low-fat-fed rats; HF, high-fat-fed rats
Phosphorylation of eNOS and myocardial NO x concentration
Since phosphorylation of eNOS is a crucial step in mediating sevoflurane-induced preconditioning, we measured myocardial Ph-eNOS expression and NO x levels.
The Ph-eNOS (normalized to total) and NO
x
concentrations were similar between two control groups (Figure 4). Sevoflurane preconditioning significantly (P < 0.05 versus LF-Control) augmented Ph-eNOS expression as well as NO
x
concentration in LF-fed rats and these increase were inhibited by AMPK inhibitor ara-A. Neither Ph-eNOS expression nor NO
x
concentration was changed in response to sevoflurane or ara-A in HF-fed rats. A-769662 administration equally increased Ph-eNOS expression and NO
x
concentration in both LH- and HF-fed rats. Changes in Ph-eNOS expression and NO
x
concentration were consistent with Ph-AMPKThr172, suggesting that eNOS-mediated NO
x
generation was related to a change in phosphorylation of AMPKThr172 in the myocardium.
Quantitative comparison of phosphorylation (Ph-) level of endothelial nitric oxide synthase (a) and nitrite and nitrate (NO
x
) generation (b) in ischemic myocardium from each group. Representative Western blots are aligned with the matching grouped data. Level of phosphorylated protein was normalized to the total protein concentration. Values are expressed as means ± SEM (n = 6 for each group). *P < 0.05 versus respective control, †
P < 0.05 versus respective LF. LF, low-fat-fed rats; HF, high-fat-fed rats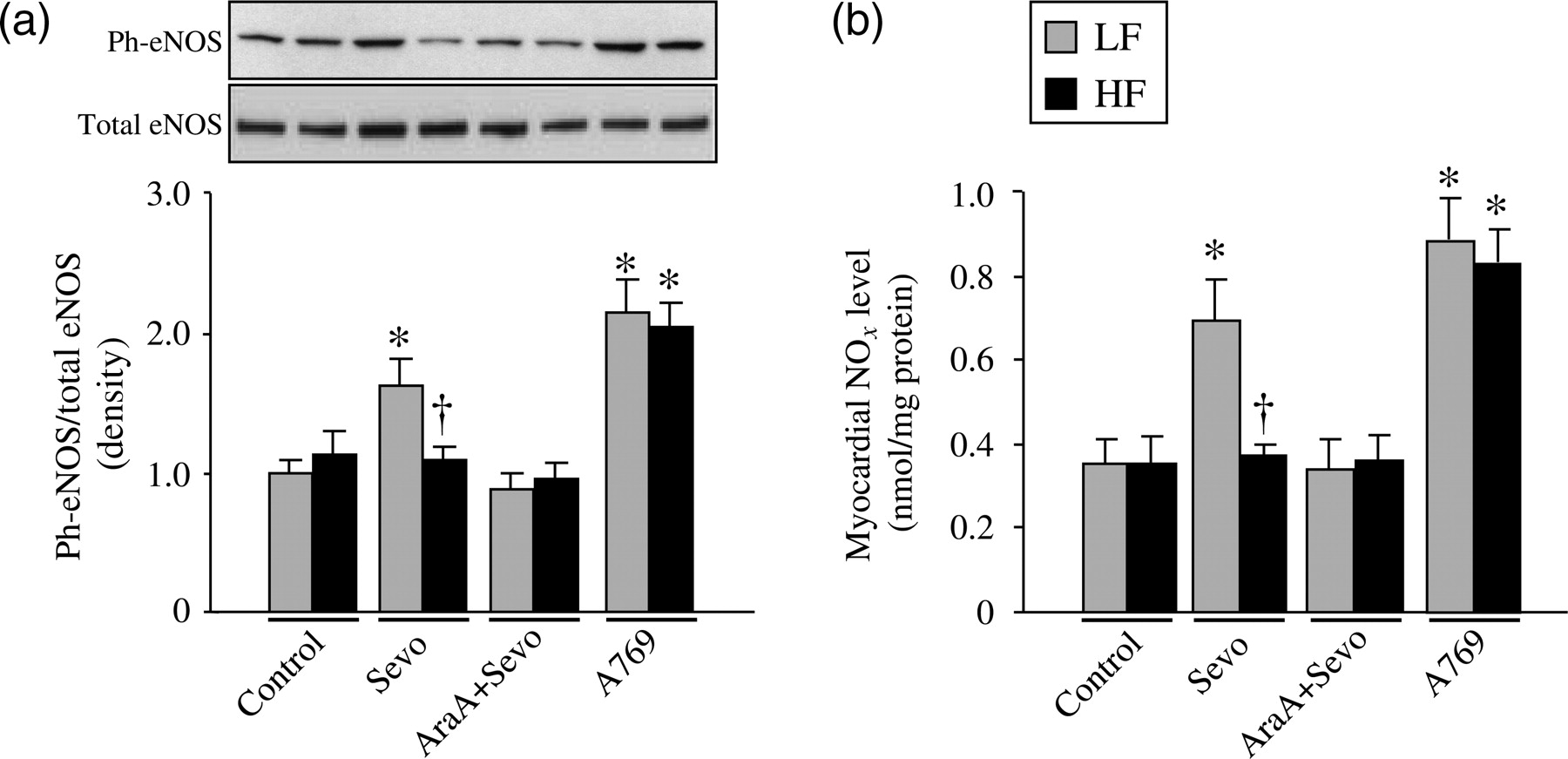
Myocardial ROS production
ROS is a necessary mediator for activation of AMPK in anesthetic preconditioning-induced cardioprotection. To determine whether HF-induced obesity influence ROS generation, we measured in situ levels of superoxide production using DHE staining.
Compared with LF-Control rats, DHE fluorescence was significantly higher in ischemic myocardium of HF-Control rats (Figure 5). Sevoflurane preconditioning enhanced the fluorescence in LF-fed rats but not in HF-fed rats. Ara-A did not alter sevoflurane-induced increase of fluorescence in LF-fed rats nor change fluorescence in HF-fed rats. Fluorescence in A-769662-treated groups was similar to their control groups.
In situ detection of reactive oxygen species production by dihydroethidium (DHE) fluorescence. (a) Representative laser confocal images from each group. Scale bar = 200 μm. (b) Quantitative comparison of DHE fluorescence in the ischemic regions of myocardium. Values are expressed as means ± SEM (n = 4 for each group). *P < 0.05 versus respective control, †
P < 0.05 versus respective LF. LF, low-fat-fed rats; HF, high-fat-fed rats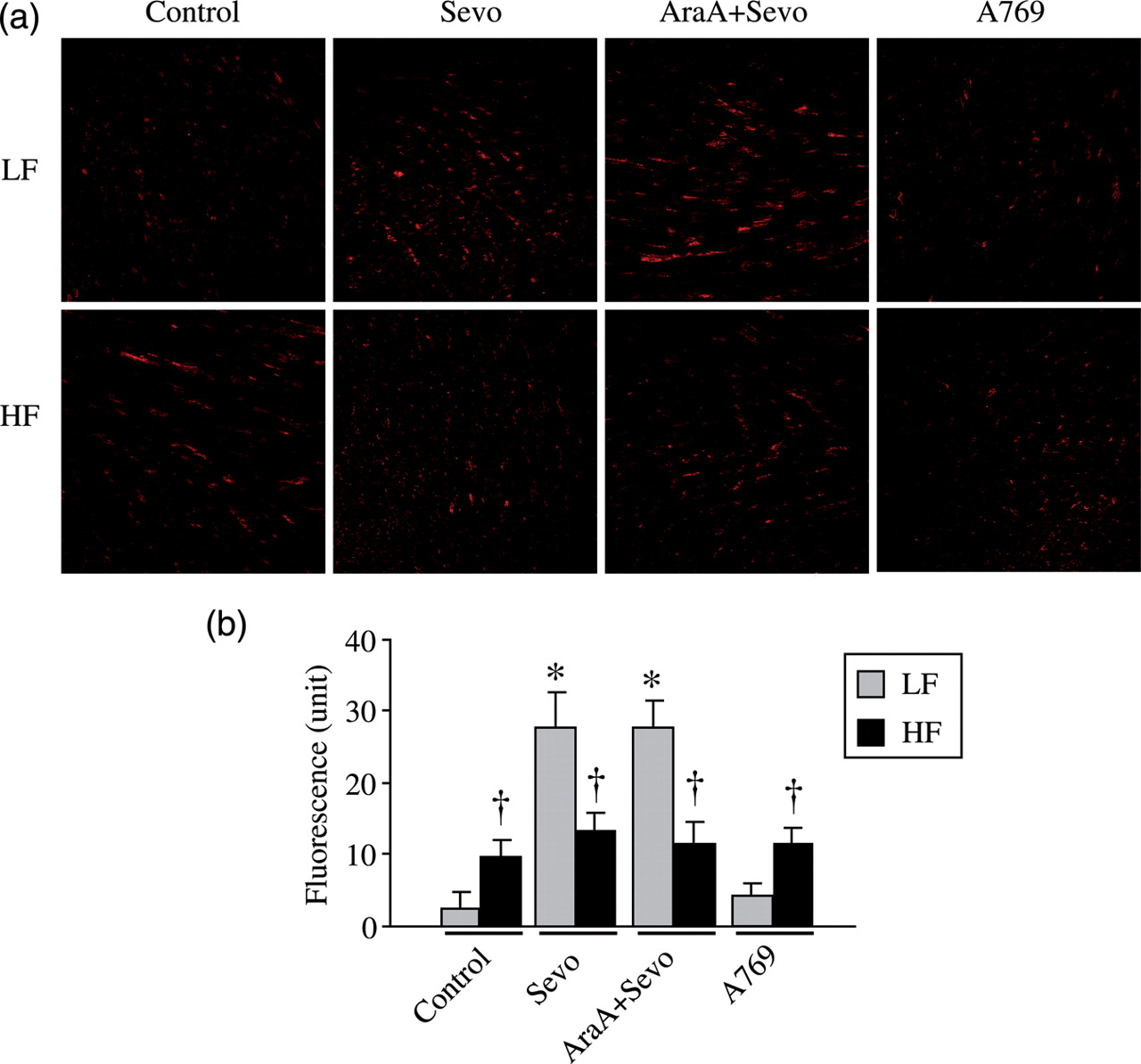
Discussion
The important new finding of this study is that high-fat diet-induced obesity suppresses anesthetic sevoflurane preconditioning-induced cardioprotective actions due to a diminished effect on ROS-mediated AMPK activation. Direct activation of AMPK by its activator reduced infarct size in both low- and high-fat diet-fed rats. To our knowledge, this is the first study to examine the cardioprotective effects of anesthetic preconditioning in obesity.
Obesity-related disorders are associated with the worse outcomes of ischemic heart diseases, but the mechanisms responsible are poorly understood. Therefore, the availability of useful animal models reflecting the human obesity syndrome is crucial for investigating the pathophysiology, and in the search for a pharmacological treatment of cardiovascular diseases in obesity. Although genetic animal models are commonly used for obesity-related studies, the gene mutations causing obesity in humans are rare, and it is generally accepted that the tremendous rise in the obesity prevalence across the globe is driven primarily by a combination of increased calorie intake and decreased physical activity. 25 The high-fat-diet-induced obese animal model has been considered to be an appropriate animal model closely mimicking the human obesity syndrome. 26 In the present study, rats fed with a high-fat diet for 12 weeks developed metabolic alterations including visceral obesity, hyperinsulinemia, hyperleptinemia and dyslipidemia. The characteristics and metabolic parameters at termination of the study are consistent with a previous report, 27 indicating that this animal model mimics the classical insulin resistance and leptin resistance features of human obesity. A previous study showed that hearts from obese Zucker rats exhibited larger infarct size following ischemia–reperfusion than those from control rats. 28 In the present study, we found that ischemia–reperfusion without preconditioning induced similar myocardial infarct size between the LF- and HF-fed rats. This result confirmed findings of an earlier study 29 demonstrating that rats fed a high-fat diet developed metabolic syndrome but did not influence myocardial infarct size. More importantly, we found that sevoflurane preconditioning reduced ischemia–reperfusion-induced infarct size in LF-fed rats but not in HF-fed rats. Our data extended the previous results and demonstrated that high-fat-diet-induced obesity inhibited sevoflurane preconditioning-induced cardioprotective actions.
Recent studies demonstrated that in normal animals, the activation of AMPK is involved in anesthetic preconditioning-induced cardioprotection. 6,30 Activation of AMPK leads to phosphorylation of eNOS and generation of NO x , which is a crucial step in mediating cardioprotection by sevoflurane preconditioning. 19 Phosphorylation of AMPK at Thr172 accounts for most of the activation of AMPK by upstream AMPK kinase. Phosphorylation of AMPK at the new site Ser485/491 was not essential for AMPK activity, but may inhibit AMPK activity. 24 It has been reported that insulin antagonizes ischemia-induced AMPK activity via activation of AMPK at Ser485/491 by protein kinase B. 24 Furthermore, high-fat-diet-induced obesity with insulin resistance inhibits the activation of AMPK signaling in skeletal muscle during exercise in mice. 16,31 We hypothesized that diet-induced obesity may inhibit the activation of AMPK signaling in the heart, resulting in impaired anesthetic preconditioning against ischemic cardiac injury. We observed that sevoflurane preconditioning in LF-fed rats significantly increased the phosphorylated state of AMPK following ischemia. Consequently, phosphorylation of eNOS was increased as well and accompanied by increased myocardial NO x production. Inhibition of AMPK activity with ara-A reversed all these increases and abolished the effects of sevoflurane preconditioning on myocardial infarct size. In contrast, sevoflurane preconditioning failed to increase the phosphorylation of AMPK and eNOS as well as myocardial NO x generation and was unable to elicit a similar cardioprotective effect in HF-fed rats. In addition, directly enhancing AMPK activation with its activator A-769662, which had been recently shown to protect the diabetic heart against ischemia–reperfusion injury, 32 significantly decreased infarct size in both LF- and HF-fed rats, indicating that the sevoflurane preconditioning induced augmented phosphorylation of AMPK and eNOS to stimulate NO x production during ischemia are functionally related to the cardioprotection. These results were consistent with our hypothesis that dysfunctional AMPK activity contributed to impaired cardioprotective actions by sevoflurane preconditioning in high-fat diet-induced obesity. Notably, the phosphorylation of AMPK was equally increased in response to A-769662 treatment between two groups, but infarct size was still higher in HF-fed rats than that in LF-fed rats, suggesting that other signaling pathways may also be involved in decreased response to sevoflurane preconditioning in high-fat-diet-induced obesity. For example, plasma level of adiponectin, an adipocytokine secreted from adipose tissue, is significantly reduced in HF-fed rats. It had been reported that decreased adiponectin concentration observed in obesity or type II diabetes was inversely correlated with the risk of myocardial infarction and contributed to increased ischemic heart disease morbidity. 33,34 Adiponectin knockout mice exhibited increased ischemia–reperfusion injury, which was ameliorated by exogenous supplementation of adiponectin. 35,36 Adiponectin might directly protect myocardium during ischemia–reperfusion independent of AMPK. 37 Thus, the reduced adiponectin concentration may also play a causative role in decreased response to sevoflurane preconditioning in high-fat-diet-induced obesity.
How does sevoflurane preconditioning fail to enhance AMPK activation in high-fat-diet-induced obesity? LF-fed rats treated with AMPK activator A-769662 exhibited a robust increase in phosphorylation of AMPK at Thr-172 when compared with LF-fed control rats. Interestingly, HF-fed rats were equally responsive to A-769662 treatment, suggesting that the AMPK itself is intact in HF-fed rats. Insulin may antagonize AMPK activity via activation of AMPK at Ser485/491 by upstream kinase LKB1. 24 To examine if the decrease in AMPK activation could be due to activation of AMPK at Ser485/491 by their major upstream kinase LKB1 in response to plasma insulin, which was elevated in HF-fed rats, we measured phosphorylation of AMPK at Ser485/491 and LKB1. We found that neither phosphorylation of AMPK at Ser485/491 nor LKB1 was altered among all experimental groups. These data suggested that dysfunctional phosphorylation of AMPK was unlikely due to the inhibitory action by activation of AMPK at Ser485/491 or upstream kinase LKB1. Generation of ROS in the context of preconditioning has been shown to be beneficial. Augmented mitochondrial ROS level in response to preconditioning has been proposed as a trigger for signaling pathways conferring cardioprotection. 23,28 It has been reported that sevoflurane-induced AMPK activation protects the heart against ischemia–reperfusion injury and this activation relies on upstream production of ROS. 10 We next examined ROS production in ischemic myocardium. DHE staining revealed that ROS production was higher in HF-fed control rats than that in LF-fed control rats, indicating a state of increased oxidative stress in HF-fed rats. Sevoflurane preconditioning induced enhanced ROS generation in LF-fed rats but failed to enhance ROS generation in HF-fed rats. This result was consistent with a published report 28 in Zucker obese rats, suggesting that an increased oxidative stress associated with insulin resistance is central to the impaired ROS generation in response to preconditioning in obese hearts. Increased oxidative stress at baseline may cause oxidative modification of key mitochondrial enzymes and/or ion channels, leading to failure of enhanced ROS generation in the mitochondria of obese hearts by preconditioning or diazoxide. 28 Interestingly, the phosphorylation of AMPK expression was similar between the two control groups, despite the basal ROS level being higher in HF-fed control rats compared with LF-fed control rats. We speculate that elevated ROS generation alone associated with insulin resistance under basal condition may not directly contribute to activation of AMPK. However, the excessive ROS generation is necessary for activation of AMPK mediated by sevoflurane preconditioning. Indeed, AMPK activator A769662 treatment did not change phosphorylation of AMPK expression, but significantly increased activation of AMPK in both groups. Taken together, these data showed that diminished AMPK activation in response to sevoflurane preconditioning is not due to deficiencies in upstream kinase function or AMPK itself, but rather a consequence of oxidative stress in HF-fed rats. Thus, the inability of sevoflurane preconditioning to increase excess ROS generation from the hearts of HF-fed rats may eliminate an essential preconditioning stimulus for activation of the AMPK signaling pathway.
A limitation should be acknowledged regarding the present study. Our results indicated that activation of AMPK exerts cardioprotective actions by increasing the expression of its downstream signaling pathway involving eNOS to stimulate NO release, which functions as both a trigger and a mediator of cardioprotection produced by preconditioning. Administration of NO donors that increase NO release prior to ischemia have been shown to protect the myocardium against ischemia–reperfusion injury. 38 However, we did not use an NO donor in the present study. Further studies are needed to examine whether NO donors, such as sodium nitroprusside, may overcome the decreased response to sevoflurane preconditioning and protect the myocardium in high-fat-diet-induced obesity.
In summary, the present study demonstrates that diet-induced obesity impairs the ability of anesthetic sevoflurane preconditioning to protect the heart against myocardial infarction. The lack of enhanced phosphorylation of AMPK by sevoflurane preconditioning might partly explain the loss of cardioprotection in this experimental model of obese rats. Targeting AMPK may be critical to elicit cardioprotection by anesthetic preconditioning in human obesity.
Footnotes
ACKNOWLEDGEMENTS
The research was supported by a Grant-In-Aid award (No. 30840075) from the National Natural Science Foundation of China (to TS).