
Abstract
Urinary trypsin inhibitor (UTI) is a serine protease inhibitor produced in the liver that inhibits the production and activation of various cytokines, notably transforming growth factor-β (TGF-β), which are associated with the progression of liver fibrosis. However, the various roles of endogenous UTI in liver fibrosis have not been examined. This study, therefore, examined the long-term effects of UTI deficiency during both steady-state conditions and thioacetamide (TA)-induced liver fibrosis. Furthermore, the effects of continuous exogenous UTI administration were examined. Analyses of liver fibrosis marker, hyaluronic acid (HA), TGF-β concentrations and histological findings at 30 weeks of age showed that homozygous UTI-knockout (KO) mice had higher HA and TGF-β concentrations than did heterozygous UTI-KO mice and wild-type mice, although there was no histological evidence of liver fibrosis in these mice. TA treatment for 20 weeks also resulted in greater HA and TGF-β levels in homozygous mice than in heterozygous and wild-type mice. Furthermore, homozygous mice had more severe liver fibrosis based on histological analyses. HA and TGF-β levels were lower in homozygous UTI-KO mice that were continuously administered UTI versus those given distilled water. These findings indicate that UTI deficiency leads to the production of HA and hepatic TGF-β and that administering exogenous UTI can ameliorate these changes. We conclude that endogenous UTI is produced in the liver to suppress the production and activation of TGF-β and that administering exogenous UTI may be therapeutically beneficial for preventing liver fibrosis.
Introduction
Urinary trypsin inhibitor (UTI) is a serine protease inhibitor derived from the degradation of pre-α-/inter-α-trypsin inhibitors synthesized in the liver 1,2 in response to biological insults such as infection, surgery, trauma and inflammation. 3 This molecule is believed to serve as an intrinsic protective factor against biological insults. In Japan, a highly purified preparation of UTI, ulinastatin, has been clinically used in the treatment of circulatory shock and pancreatitis. 3,4 Research using the rat hepatic ischemia–reperfusion injury model found that exogenous administration of UTI significantly reduced liver damage. 5–8 However, little is known about the physiological significance of endogenous UTI for the liver, the organ that secretes this protease inhibitor. 1,2,9
During hepatopathy induced by chronic inflammation (i.e. chronic hepatitis), damaged tissue is repaired by accumulated connective tissue, which causes the liver to ultimately become cirrhotic. As fibrosis is the most important factor in the onset of both carcinogenesis and hepatic failure, one important prophylactic and therapeutic strategy is to control the progression of hepatic fibrosis. 10,11 The progression of liver fibrosis has been shown to involve transforming growth factor-β (TGF-β), released from activated stellate cells, as well as plasmin, kallikrein and other liver serine proteases that activate stellate cells. 12–14
Extensive investigation has been successfully revealing the intrinsic protective roles of UTI. One of the key actions of UTI identified, to date, is the suppression of the production of tumor necrosis factor-α, interleukin-1β and other inflammatory cytokines. 15,16 UTI has been recently reported to inhibit radiation-induced lung fibrosis by potentially suppressing TGF-β synthesis. 17 Other important effects of UTI include the inhibition of the release of various proteases by stabilizing the lysosomal membrane. 18 Past research has identified that the target proteases of UTI include trypsin as well as serine proteases involved in the activation of TGF-β. 8,19–21
These findings led us to hypothesize that UTI endogenously produced in the liver inhibits the development of liver fibrosis resulting from viral hepatitis and other causes by suppressing the production and activation of TGF-β.
In this study, we employed UTI-knockout (KO) mice to investigate the physiological significance of endogenously produced UTI in the prevention of liver fibrosis. We compared the differences among homozygous UTI-KO, heterozygous UTI-KO and wild-type mice in (1) age-related hepatic changes and (2) the development of liver fibrosis induced by the potent hepatotoxin, thioacetamide (TA). 22
Materials and methods
Animals
The UTI-KO mice used in this study (provided by the Research Center, Mochida Pharmaceutical Co, Ltd, Shizuoka, Japan) were generated by replacing exons 8 and 9 of the UTI gene with the neomycin-resistance gene. 23 Specifically, they were produced as follows: 129/Ola-derived embryonic stem (ES) cells were first transfected with the gene-targeting construct containing the neomycin-resistance gene, and gene-targeted ES cells were selected by screening for neomycin resistance. The selected ES cells were injected into host C57BL/6 blastocysts, and the blastocysts were transferred to the uterus of pseudo-pregnant ICR mice to produce chimeric offspring with a mosaic (black and white) coat color. Chimeras were then mated with C57BL/6 mice to yield white mice heterozygous for the gene KO (the coat color was the same as that for the original ES cell line), and these heterozygous mice were mated to finally produce homozygous KO mice. Western blot analysis was used to confirm the constitutive KO. The experimental protocols were approved by the Animal Care Committee of Asahikawa Medical University and were in accordance with the National Institute of Health's ‘Guide for the Care and Use of Laboratory Animals’.
Treatments and sample collection
Ten-week-old homozygous and heterozygous UTI-KO and wild-type mice were divided into two groups (n = 10 each), and were orally administered either TA (purchased from Sigma Chemical Japan, Co, Tokyo, Japan) or vehicle. Animals were killed 20 weeks after the treatment (at 30 weeks of age). Blood samples were collected from all mice, and the serum was separated and stored frozen. Liver lobes were extirpated, and tissues were stored frozen or fixed with 4% paraformaldehyde. TA was mixed in drinking water (300 mg/L) for oral administration in accordance with a previous report. 24 Two groups of five homozygous UTI-KO mice were randomly selected at 26 weeks of age, and underwent surgery under sodium pentobarbital anesthesia (50 mg/kg, intraperitoneally) for implantation of a mini-osmotic pump (Model 2004; Alza, Palo Alto, CA, USA; flow rate of 0.25 μL/h, maximum total volume of 200 μL). This pump was used to continuously deliver either ulinastatin, UTI agent (10,000 units in 200 μL solution purchased from Mochida Pharmaceutical Co, Ltd) or vehicle (distilled water) for four weeks. The amount of ulinastatin administered per day was calculated at 357 units, resulting in 12,000 units/kg over the four-week period. Both groups of mice (treated with ulinastatin and control) were killed at 30 weeks of age for measurements of serum liver fibrosis markers and histological examination of liver tissues.
Assessment of liver fibrosis
Serum levels of hyaluronic acid (HA), a fibrosis marker, were determined at a contract laboratory (SRL Hokkaido, Inc, Hokkaido, Japan). For histological evaluation of liver damage, tissue samples were stained with hematoxylin–eosin and graded according to the criteria reported earlier: 0, no fibrosis; +1, slight fibrosis located in the central liver lobule; +2, moderate wide central fibrosis; +3, severe fibrosis extending to the edge of the liver lobule; and +4, liver cirrhosis. 25 Separately, liver sections were stained with 0.1% Sirius red and 0.1% fast green counterstain (Sigma Chemical Japan) to visualize collagen fibers. Images obtained with a Nikon microscope and DS Camera Control Unit (DS-L1; Nikon Co, Tokyo, Japan) were quantitatively analyzed according to previously reported procedures utilizing NIH Image software (Version 1.62; National Institutes of Health, Bethesda, MD, USA) and Adobe® Photoshop image processing software (Adobe Systems Inc, Japan, Tokyo, Japan). 26–28 Alpha smooth muscle actin (α-SMA) has been advocated as an effective marker for activated stellate cells. We stained activated stellate cells as per the previously described polymer immunocomplex method using mouse monoclonal anti-α-SMA antibody (Dako Japan, Tokyo, Japan). 29 Immunohistochemical staining of mouse tissue with mouse monoclonal antibody often results in undesirable background staining because antimouse immunoglobulin secondary antibodies react with endogenously produced mouse immunoglobulin present in the mouse tissue specimen. The polymer immunocomplex staining method eliminates the adverse influence of endogenous mouse immunoglobulin and provides highly specific staining. This method employs a new type of dextran polymer conjugated with secondary antibody having antimouse immunoglobulin specificity (EnVision; Dako Japan; 100–500 μL/plate). Polymer immunocomplex is formed by mixing this reagent with mouse monoclonal primary antibody and then with normal immunoglobulin (blocking agent for unbound secondary antibody) at room temperature for one hour. The resulting polymer immunocomplex was added to the mouse tissue sections, which were left standing at 4°C overnight. 3,3′-Diaminobenzidine was used to develop the immunostain. The dextran polymer-based detection reagent (EnVision or Mouse), normal mouse serum (Code no. X0910), color development substrate (3,3′-diaminobenzidine) and other materials used for immunohistochemical staining in this study were purchased from Dako Japan. Visual fields were randomly chosen from the stained images, and classified into four groups according to the average proportion of positive cells as described elsewhere 21 : 0, no positive cells; 1, positive cells <1/3; 2, positive cells <2/3; and 3, positive cells >2/3. Each plate's score was calculated by taking the mean value of 20 randomly selected visual fields. 25
Reverse transcription polymerase chain reaction
Hepatic mRNA samples were extracted using Sepazol RNA-I (Nacalai Tesque, Kyoto, Japan), and the TGF-β expression in liver tissue was determined by reverse transcription polymerase chain reaction (RT-PCR) using Takara RNA PCR Kit (AMV) Version 3.0 (Takara Bio Inc, Tokyo, Japan). The sequences for the upstream and downstream PCR primers for TGF-β (PCR product size, 353 bp) were 5′ GCC TGA GTG GCT GTC TTT TGA CG 3′ and 5′ ACT TCC AAC CCA GGT CCT TC 3′, respectively. The upstream and downstream PCR primers for β-actin (PCR product size, 112 bp) had the following sequences, respectively: 5′ TCA GCA AGC AGG AGT ACG ATG A 3′ and 5′ TGC GCA AGT TAG GTT TTG TCA A 3′. The resulting PCR product bands were quantitatively analyzed by using the NIH Image and Adobe® Photoshop software programs.
Statistics
Data are represented as means ± SD. Repeated measures analysis of variance was used for data analysis, and Duncan's post hoc analysis was performed for multiple comparisons. Mann–Whitney U test was used for two-group comparison. P values <0.05 were considered statistically significant.
Results
The effect of the absence of endogenous UTI on the liver
Liver tissue samples taken from homozygous UTI-KO, heterozygous UTI-KO and wild-type mice were examined by gross observation (Figure 1), hematoxylin–eosin staining (Table 2), Sirius red staining (Figure 2 and Table 3) and α-SMA immunohistochemical staining (Table 4) at 30 weeks of age. None of the groups demonstrated liver fibrosis by light microscopic examination. The serum levels of the fibrosis marker HA were compared among the three groups of mice (Table 1). The homozygous UTI-KO group had a higher level of HA than the other groups. The wild-type and heterozygous UTI-KO groups had very low levels of hepatic TGF-β mRNA expression. The homozygous UTI-KO group demonstrated a higher level of hepatic TGF-β mRNA expression than the other two groups. Quantitative RT-PCR analysis demonstrated a significantly higher increase in hepatic TGF-β mRNA expression for the homozygous UTI-KO group than the other groups (Figure 3a).
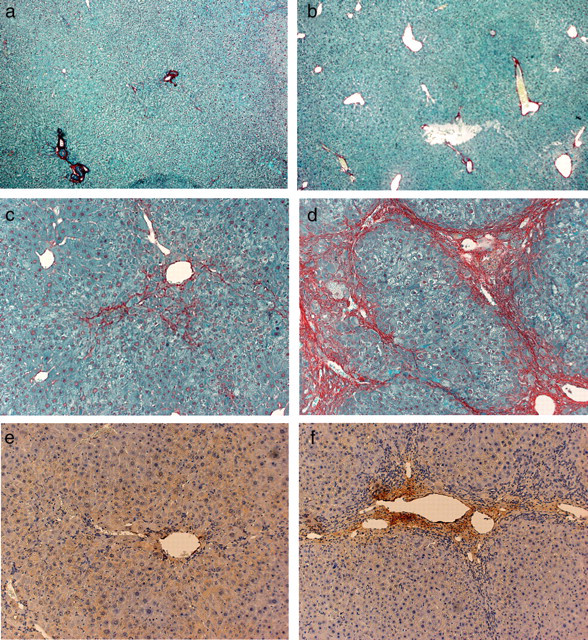
Typical macroscopic presentations of the liver from UTI-knockout (KO) and wild-type mice, treated with or without 20-week administration of thioacetamide. Untreated wild-type and homozygous UTI-KO mouse received no drug treatment. The remaining mouse received 300 mg/L thioacetamide (TA) in their drinking water for the final 20 weeks of the experiment. (a) Untreated wild-type mouse, 30 weeks of age. (b) Untreated homozygous UTI-KO mouse, 30 weeks of age. (c) Treated wild-type mouse, 30 weeks of age. (d) Treated homozygous UTI-KO mouse, 30 weeks of age. UTI, urinary trypsin inhibitor Typical histological presentations of the liver from UTI-knockout (KO) and wild-type mice, treated with or without 20-week administration of thioacetamide (TA). Untreated wild-type and homozygous UTI-KO mouse received no drug treatment. The remaining mouse received 300 mg/L TA in their drinking water for the final 20 weeks of the experiment. The sections were stained with Sirius red to detect the extracellular matrix and collagen. Activated hepatic stellate cells were detected by immunohistochemistry with a monoclonal antibody for alpha smooth muscle actin (α-SMA). (a) Untreated wild-type mouse, 30 weeks of age, Sirius red staining (×40). (b) Untreated homozygous UTI-KO mouse, 30 weeks of age, Sirius red staining (×40). (c) Treated wild-type mouse, 30 weeks of age, Sirius red staining (×100). (d) Treated homozygous UTI-KO mouse, 30 weeks of age, Sirius red staining (×100). (e) Treated wild-type mouse, 30 weeks of age, α-SMA immunohistochemical staining (×100). (f) Treated homozygous UTI-KO mouse, 30 weeks of age, α-SMA immunohistochemical staining (×100). UTI, urinary trypsin inhibitor Typical presentations of polymerase chain reaction products for TGF-β mRNA extracted from UTI-knockout (KO) and wild-type mice. (a) Wild, wild-type mouse, 30 weeks of age; hetero, heterozygous UTI-KO mouse, 30 weeks of age; homo, homozygous UTI-KO mouse, 30 weeks of age. *P < 0.01 versus wild and hetero. (b) Two groups of five homozygous UTI-KO mice received continuous intraperitoneal administration of either ulinastatin, UTI agent (12,000 units/kg/d) or vehicle (distilled water) for four weeks using a mini-osmotic pump. Vehicle + homo, homozygous UTI-KO mouse treated with vehicle; UTI + homo, homozygous UTI-KO mouse treated with ulinastatin. *P < 0.05 versus vehicle + homo. (c) The mice, 30 weeks of age, received 300 mg/L thioacetamide (TA) in their drinking water for the final 20 weeks of the experiment. TA + wild, wild-type mouse; TA + hetero, heterozygous UTI-KO mouse; TA + homo, homozygous UTI-KO mouse. *P < 0.05 versus TA + wild and TA + hetero. AU: Arbitrary units denote the band density ratio of the target gene relative to β-actin. Data are represented as means ± SD (n = 5). UTI, urinary trypsin inhibitor; TGF-β, transforming growth factor-β
Serum hyaluronic acid (HA) levels UTI, urinary trypsin inhibitor; Wild, wild-type mouse, 30 weeks of age; hetero, heterozygous UTI-knockout (KO) mouse, 30 weeks of age; homo, homozygous UTI-KO mouse, 30 weeks of age; TA + wild, wild-type mouse, 30 weeks of age, after 20-week administration of thioacetamide (TA); TA + hetero, heterozygous UTI-KO mouse, 30 weeks of age, after 20-week administration of TA; TA + homo, homozygous UTI-KO mouse, 30 weeks of age, after 20-week of administration of TA; ulinastatin + homo, homozygous UTI-KO mouse with four-week continuous intraperitoneal administration of ulinastatin; vehicle + homo, homozygous UTI-KO mouse with four-week continuous intraperitoneal administration of distilled water. Data are represented as mean ± SD. The column headed by ‘n’ indicates the numbers of mice tested *P < 0.01 versus wild and hetero groups, †
P < 0.01 versus wild group, ‡
P < 0.01 versus hetero group, §
P < 0.01 versus homo group, ||
P < 0.01 versus TA + wild and TA + hetero groups and **P < 0.05 versus vehicle + homo group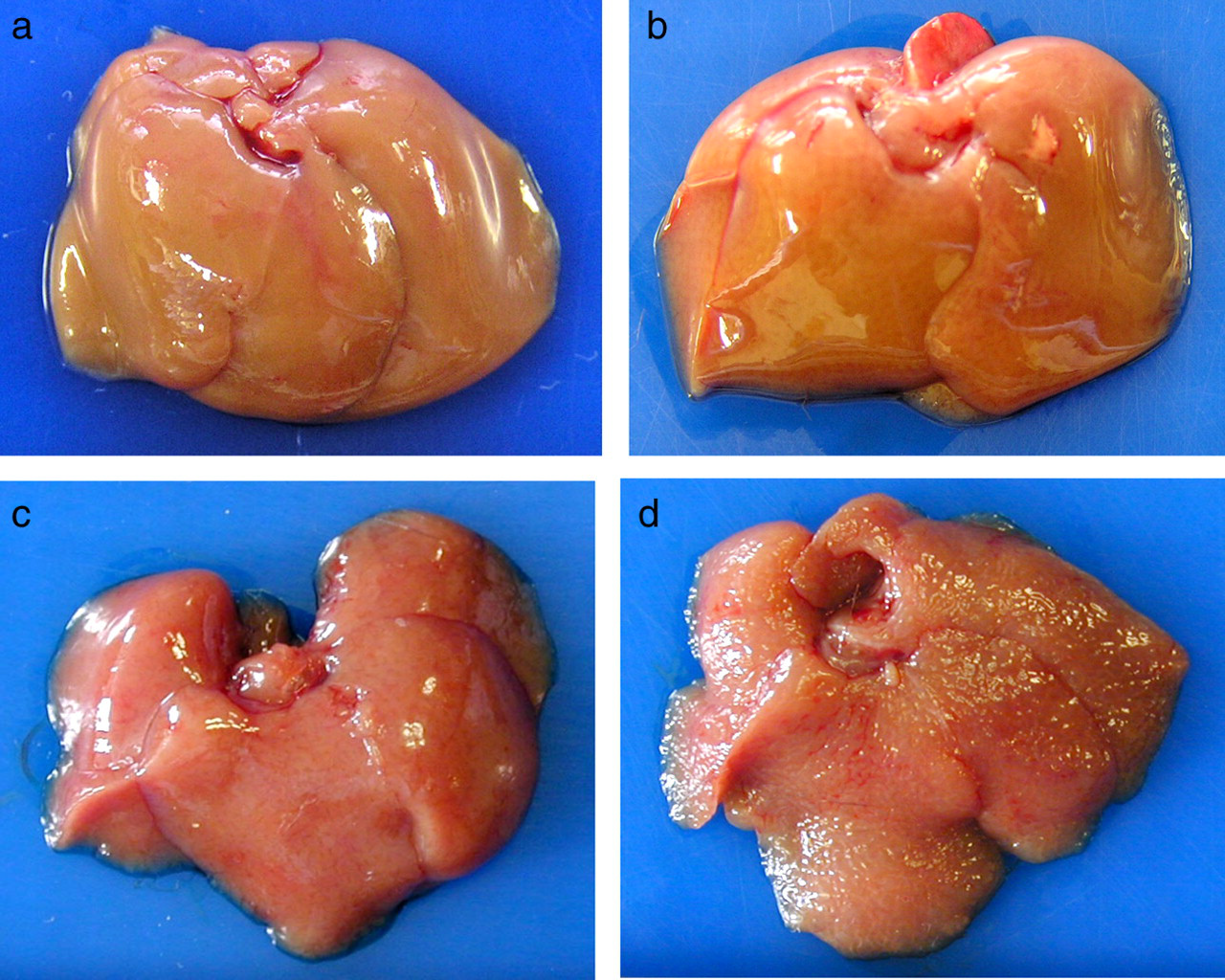

The effect of continuous UTI administration on the liver
Homozygous UTI-KO mice administered ulinastatin demonstrated a significantly lower level of serum HA than did the control group of homozygous UTI-KO mice administered distilled water (Table 1). In addition, quantitative RT-PCR analysis showed that TGF-β expression was significantly suppressed in homozygous UTI-KO mice continuously administered ulinastatin than in homozygous UTI-KO mice administered vehicle only (Figure 3b).
Development of TA-induced liver fibrosis
Pathological grading of liver fibrosis based on hematoxylin–eosin-stained samples
UTI, urinary trypsin inhibitor; Wild, wild-type mouse, 30 weeks of age; hetero, heterozygous UTI-knockout (KO) mouse, 30 weeks of age; homo, homozygous UTI-KO mouse, 30 weeks of age; TA + wild, wild-type mouse, 30 weeks of age, after 20-week administration of thioacetamide (TA); TA + hetero, heterozygous UTI-KO mouse, 30 weeks of age, after 20-week administration of TA; TA + homo, homozygous UTI-KO mouse, 30 weeks of age, after 20-week of administration of TA. Data are represented as mean ± SD. The column headed by ‘n’ indicates the numbers of mice tested
*P < 0.01 versus TA + wild and TA + hetero groups
Pathological grading of liver fibrosis based on Sirius red-stained samples
UTI, urinary trypsin inhibitor; Wild, wild-type mouse, 30 weeks of age; hetero, heterozygous UTI-knockout (KO) mouse, 30 weeks of age; homo, homozygous UTI-KO mouse, 30 weeks of age; TA + wild, wild-type mouse, 30 weeks of age, after 20-week administration of thioacetamide (TA); TA + hetero, heterozygous UTI-KO mouse, 30 weeks of age, after 20-week administration of TA; TA + homo, homozygous UTI-KO mouse, 30 weeks of age, after 20-week administration of TA. Data are represented as mean ± SD. The column headed by ‘n’ indicates the numbers of mice tested
*P < 0.01 versus wild, hetero and homo groups, respectively. † P < 0.01 versus TA + wild and TA + hetero groups
Evaluation of the activation of stellate cells based on immunohistochemical staining of alpha smooth muscle actin
UTI, urinary trypsin inhibitor; Wild, wild-type mouse, 30 weeks of age; hetero, heterozygous UTI-knockout (KO) mouse, 30 weeks of age; homo, homozygous UTI-KO mouse, 30 weeks of age; TA + wild, wild-type mouse, 30 weeks of age, after 20-week administration of thioacetamide (TA); TA + hetero, heterozygous UTI-KO mouse, 30 weeks of age, after 20-week administration of TA; TA + homo, homozygous UTI-KO mouse, 30 weeks of age, after 20-week of administration of TA. Data are represented as mean ± SD. The column headed by ‘n’ indicates the numbers of mice tested
*P < 0.01 versus wild, hetero and homo groups, respectively. † P < 0.01 versus TA + wild and TA + hetero groups
Discussion
TGF-β is produced in its latent form by stellate cells, Kupffer cells and platelets, and is converted to its active form by proteases. 12,30–34 It has been clearly documented that the conversion of latent to active TGF-β in the liver requires a serine protease. 12,29 In particular, the serine protease plasmin has been shown to play a key role in TGF-β activation during the development of liver fibrosis and cirrhosis. Another common protease, kallikrein, is known to activate TGF-β when liver regeneration is impaired. 12,35 We maintain that endogenous UTI suppresses TGF-β activation and liver fibrosis, independent of hepatic pathology type, because it inhibits a large number of serine proteases and other proteases. Analysis of the individual effects of UTI on these proteases was, however, beyond the scope of the present study. Such analysis will be the focus of future studies.
In the present study, we employed UTI-KO mice and investigated the various roles of endogenous UTI in the process of liver fibrosis. To the best of the authors’ knowledge, the present study was the first to investigate the mechanisms of liver fibrosis in endogenous UTI-KO mice. The use of genetically engineered animals is an extremely useful method for investigating the effects of endogenous factors possibly involved in the development of liver fibrosis. 27,36 Our interest focused on the physiological functions of endogenous UTI in the liver, in particular its suppressive potential against liver fibrosis.
Various nutrients absorbed through the intestinal mucous membrane have been documented to reach the liver via the portal vein. A small amount of bacterial toxins from the decomposition of dead bacteria in the intestinal flora has been reported to enter the liver via the portal vein as well. 37–39 Moreover, cirrhotic patients are continuously exposed to a low concentration of endotoxin. 40,41 These facts suggest that the liver is at a constant risk of being injured by factors of external and internal origin, even in healthy individuals. Such injuries can potentially cause liver fibrosis. 11,42 In this study, concentrations of hepatic TGF-β and serum hyaluronic acid (a surrogate measure for liver fibrosis) were elevated in the endogenous UTI-KO mice and were suppressed by administration of exogenous UTI, ulinastatin. Moreover, in our experiment with TA-induced liver fibrosis using KO mice unable to produce UTI in the liver, homozygous UTI-KO mice showed a significantly higher degree of liver fibrosis than did heterozygous UTI-KO and wild-type mice. These results strongly suggest that the liver possesses a self-protective mechanism whereby it inhibits the development of fibrosis through endogenous production of UTI that suppresses the production and protease-induced activation of TGF-β. However, our study results require careful scrutiny, as they failed to demonstrate that the biological profiles of all other endogenous substances involved in liver fibrosis were similar between the genetically engineered and wild-type mice. TGF-β induces an increase in hepatic collagen α1(I) expression; 7,10 therefore, verification of hepatic collagen α1(I) expression in homozygous UTI-KO mice would verily provide strong evidence; however, we have not performed this verification yet. Another possibility is an unidentified fibrosis-suppression mechanism acting in a fail-safe manner, thus preventing the development of fibrosis in UTI-KO mice. These possible mechanisms merit future research.
TA-induced murine liver fibrosis has been shown to present with pathological characteristics similar to chronic hepatitis C infection and liver cirrhosis in humans. 24,43,44 One of the limitations of the present study was that we did not demonstrate the antifibrotic effect of exogenous UTI in TA-induced liver fibrosis in UTI-KO mice. A previous study using a rat model of swine serum-induced liver fibrosis found that the protease inhibitor, camostat mesilate, had inhibition potential similar to that of UTI in its suppression of plasmin-dependent TGF-β activation, and demonstrated that this protease inhibitor was a promising drug candidate for the treatment of liver fibrosis. 14
In preliminary experiments using the mini-osmotic pump system in TA-treated homogenous UTI-KO mice, we failed to demonstrate that TA-induced TGF-β upregulation and liver fibrosis progression were suppressed by continuous infusion of ulinastatin. Since the plasma half-life of UTI is a mere 35 min, 4 a larger dose of ulinastatin over a longer period by repeated or continuous administration was possibly required to establish UTI's antifibrotic action. Such dose adjustment is one of the multiple difficulties that we must address before we can succeed in applying ulinastatin to the clinical treatment of liver fibrosis.
UTI is covalently linked to one or two polypeptides, referred to as heavy chain (HC) 1, HC 2, and HC3, via a chondroitin sulfate chain. 45 Homogenous UTI-KO mice showed no expression of UTI as well as HCs. 23 The HCs are also known as serum-derived hyaluronan-associated proteins (SHAPs). 46 SHAPs covalently bind to hyaluronan to form the SHAP–HA complex, which is associated with many inflammatory diseases. 47 Some researchers suggest that the SHAP–HA complex level is a better indicator for the progression of the stages of liver fibrosis in patients. 48 On the other hand, the SHAP–HA complex can also inhibit the development of inflammation. 49 Thus, our results of this study may provide an incentive for evaluating the effect of HC deficiency in UTI-KO mice on inflammation and liver fibrosis in future studies.
In conclusion, our results provide evidence for the potential that UTI endogenously released from the liver inhibits TGF-β production as well as proteases involved in TGF-β activation, thereby modulating the process of liver fibrosis.
Footnotes
Acknowledgements
We thank Mr Yoshiyasu Satake for his excellent technical assistance. The assistance of Miss Keiko Lee with manuscript preparation is gratefully acknowledged. This study was supported in part by a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (16659346).