
Abstract
Aldosterone (Aldo) is an important active hormone in the renin–angiotensin–aldosterone system and plays a vital role in the development of hypertension, heart failure and other cardiovascular diseases. We aimed to explore the role of endogenous Aldo in aortic calcification in rats. We induced arterial calcification in rats by intramuscular administration of vitamin D3 plus oral nicotine (VDN) and determined calcium content, 45Ca2+ accumulation and activity of alkaline phosphatase (ALP). The mRNA level of osteopontin (OPN) was measured by semi-quantitative reverse transcriptase polymerase chain reaction. Deposition of collagen in the aorta wall was measured by Sirius red staining. The content of angiotensin II (Ang II) and Aldo in plasma and myocardial and vascular tissue was determined by radioimmunoassay. In rats with VDN treatment, von Kossa staining showed calcification in vascular smooth muscle cells and extracellular matrix, and the content of calcium in calcified arteries was 5.8-fold of that in control arteries (P < 0.01). The accumulation of 45Ca2+ and activity of ALP in calcified aortic tissue was three- and 2.5-fold, respectively, that in control tissue (P < 0.01). The mRNA expression of OPN was significantly higher, by 58%, in calcified than control tissue (P < 0.01). Vascular fibrosis was greater in rats with calcified tissue than in control rats. The level of Ang II and Aldo was 58% and 80% higher, respectively, in calcified than control tissue (both P < 0.01). The changes could be significantly improved by treatment with captopril, an angiotensin-converting enzyme inhibitor, and the Aldo receptor antagonist spironolactone. These results suggest that Aldo is an endogenous bioactive factor involved in vascular calcification.
Introduction
Vascular calcification (VC) is a basic pathological process generally associated with atherosclerosis, hypertension, diabetes, chronic kidney disease and aging. In calcified blood vessels, the decrease in vasodilator function and increase in stiffness leads to thrombosis and plaque rupture, an important risk factor of cardiovascular events. VC is found in 80% of vascular injuries and 90% of patients with coronary artery disease. 1,2 VC was previously considered a passive degenerative process. Recent evidence, especially from molecular biology research, indicates that VC is a highly regulated process controlled by cells and similar to the initially adjustable processes of osteogenesis and osteoporosis. 3,4
No clinically effective prevention and treatment measures exist for VC. Recent studies demonstrated that paracrine/autocrine factors are involved in maintaining circulatory homeostasis and mediate the pathogenesis of VC. Among these factors, vasoactive peptides such as adrenomedullin, ghrelin, parathyroid hormone-related peptide and C-type natriuretic peptide, as well as gaseous signal molecules such as nitric oxide (NO), CO and H2S, are endogenous inhibitory factors in VC. However, associated factors endothelin and angiotensin II (Ang II) have opposite effects. 5–10 Thus, investigation of endogenous bioactive substances may help reveal new mechanisms and therapeutic strategies for VC.
The cardiovascular tissue renin–angiotensin–aldosterone system (RAAS) acts in a paracrine/autocrine manner and is of great significance in maintaining homeostasis in cardiovascular function and structure. This system has an important regulatory role in the long-term regulation of blood pressure and in diabetic nephropathy and especially atherosclerosis. 11 Aldosterone (Aldo) is an important active hormone in the RAAS system and is the major component in adrenal mineralocorticoids. Aldo causes retention of sodium water and can promote collagen deposition and fibrosis, thus leading to fibrosis and structural re-modeling of the heart, blood vessels and other organs. It can enhance the vascular response to norepinephrine and play an important role in the development of hypertension, heart failure and other cardiovascular diseases. 12 Ang II and Aldo in coronary artery smooth muscle cells regulate the gene transcription of alkaline phosphatase (ALP) by activating mineralocorticoid receptors (MRs). 13 Aldo also promotes the calcification of in vitro-cultured calcifying vascular cells by activating MRs. 14 However, the function and mechanism of Aldo in the calcification of vascular tissue in animals are unclear.
Excessive amounts of vitamin D3 can increase the intestinal absorption of calcium and increase the concentration of calcium in the blood. In addition, vitamin D3 can degrade the elastic fiber of blood vessel walls in rats and rabbits, promote the deposition of calcium and phosphorus, and activate the vitamin D receptor in smooth muscle cells, thereby increasing the intake of calcium and leading to intracellular calcium overload. Excessive amounts of vitamin D3 can promote the expression of calcification-related genes, thus contributing to calcification. Nicotine can enhance the effects of vitamin D3. The two combined can cause calcium overload in cardiovascular tissues and deposition of calcium in artery media and elastic fibers, eventually leading to VC. 15
We hypothesized that Aldo is an endogenous bioactive factor and takes part in VC. We used a rat VC model induced by vitamin D3 plus nicotine (VDN) to investigate the changes in endogenous Aldo level and the impact of captopril, an angiotensin-converting enzyme inhibitor (ACEI), and spironolactone, an Aldo receptor antagonist on VC. Aldo participated in VC in rats; the generation of Ang II and Aldo was increased in VDN-induced VC, and treatment with ACEI and Aldo receptor antagonist improved cardiac function in induced VC.
Materials and methods
Materials
All animal husbandry and experimental procedures were performed in accordance with the Animal Management Ordinance of the Chinese Ministry of Health (document no. 55, 2001) and the animal experiment standards approved by the Animal Management Committee of Peking University Health Science Center.
Adult male Sprague–Dawley (SD) rats (180–200 g) were obtained from the Department of Laboratory Animal Science of Peking University Health Science Center. Nicotine, vitamin D3, spironolactone and captopril were from Sigma (St Louis, MO, USA); Dulbecco's modified Eagle's medium was from Gibco (Gaithersburg, MD, USA); 45CaCl2 was from the New England Nuclear Corp (Boston, MA, USA), with a radioactive concentration of 15 mCi/mL and specific activity of 18.373 mCi/mg (679.80 Mbq/mg); Moloney-murine-leukaemia virus (MMLV) reverse transcriptase, Taq DNA polymerase, RNasin and oligo (dT) 15 primers were from Promega (Madison, WI, USA); and reverse transcriptase polymerase chain reaction (RT-PCR) primers for osteopontin (OPN) were synthesized by Beijing Parkson Corp (Beijing, China). The ALP detection kit was from Beijing Furui Co (Beijing, China) and Ang II and Aldo radioimmunoassay (RIA) kits were from Beijing East Asia Institute of Immunological Technology. Other chemicals and reagents were of analytical grade.
Preparation of VC model and experimental groups
The VC rat model was prepared as described 5,15 with minor modification. Briefly, 32 male SD rats were randomly divided into four groups (n = 8) for treatment: (1) VDN: VC induced by intramuscular injection of vitamin D3 (300,000 IU/kg) and nicotine (25 mg/kg) dissolved in peanut oil administered by gavage and repeated once nine hours later; (2) VC plus captopril (VDN + captopril): VC, then captopril (100 mg/kg/d) administered by gavage for four weeks; (3) VC plus spironolactone (VDN + spironolactone): VC, then spironolactone (100 mg/kg/d) administered by gavage for four weeks; and (4) control: intramuscular injection of normal saline and peanut oil administered by gavage alone.
Rats of all groups were fed conventionally and periodically reviewed for weight, and doses were adjusted until four weeks after VC treatment. At the last administration, all animals fasted but had free access to water. Then, 12 h later, rats were anesthetized by intraperitoneal injection of 30 mg/kg sodium pentobarbital; the catheter system was filled with heparin to prevent coagulation of blood. Artery and cardiac catheters were inserted in the left femoral artery and right carotid artery. The heart rate (HR), arterial blood pressure and left ventricular (LV) pressure – LV end systolic pressure (LVESP), LV end diastolic pressure (LVEDP) and ±LV dp/dt – were recorded by use of Powerlab (ADInstruments, Sydney, Australia). Blood was taken from the femoral artery at the end of the experiment. Blood samples were mixed with 1 mg/mL ethylenediaminetetraacetic acid (EDTA)-Na2 and 500 kallikrein inactivator units/mL of aprotinin. Plasma was obtained by centrifugation at 1600 g for 15 min at 4°C and preserved at −70°C. Rats were killed, and the heart and all thoracic and abdominal arteries were collected. After flushing the blood vessel with cold phosphate-buffered saline (PBS), 5-mm aorta tissue was collected and fixed with 10% formalin for histology; the remaining artery tissue was used for other experiments.
Von Kossa staining of the artery wall
We performed von Kossa calcium staining as we described previously. 16 A 1-cm segment of aorta was excised and fixed with 10% phosphate-buffered neutral formalin (pH 7.4, 0.1 mol/L) for 24 h, then samples were dehydrated in a graded series of ethanol solution and embedded in paraffin. Sections of aortic tissue 6-μm thick were cut, and one slide was stained with hematoxylin and eosin. Other slides were deparaffinized, dehydrated and then treated with 5% AgNO3 for 30 min. Specimens were then counterstained with safranine (red staining) and examined under a light microscope.
Sirius red staining of arteries for collagen content
A 0.5-cm segment of vascular tissue was fixed with 4% paraformaldehyde, embedded in paraffin for slicing, with slice thickness 7 μm, then underwent Sirius red staining for determining vascular collagen. Data were analyzed by polarization microscopy (Leica, Solms, Germany).
Measurement of calcium content in aorta
Calcium content in the aorta was determined as described. 16 Aortic tissue (about 10 mg) was dissolved in HNO3 and diluted with a blank solution (27 nmol/L KCl, 27 μmol/L LaCl3). The calcium content was measured by atomic absorption spectrophotometry at 422.7 nm (novAA 300; Analytik, Jena AG, Germany).
Measurement of 45Ca2+ accumulation in aortas
45Ca2+ accumulation in aortas was as described 16 with minor modification. Aortic tissue (about 20 mg) was sliced and incubated in 1 mL Krebs–Henseleit (K–H) solution (in mmol/L: 118 NaCl, 4.7 KCl, 1.3 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3 and 11 glucose; pH 7.2) with 37 kBq/mL 45CaCl2. The reaction was stopped by the addition of ice-cold K–H solution. The tissue was dissolved and protein content was determined by the Bradford method. 45Ca2+ radioactivity was measured by β-scintillation counting (LS 6500; Beckman Coulter, Fullerton, CA, USA).
Measurement of ALP activity in aorta
Approximately 10 mg (1 cm) abdominal aortic tissue was incubated with isotonic PBS (1:10, w/v) to prepare tissue homogenates. After centrifugation for 10 min at 4°C, 1600 g, the supernatant was collected and ALP protein content was determined by the Bradford method. ALP activity was determined by the use of an ALP detection kit.
Radioimmunoassay of Ang II and Aldo content in plasma and arteries
In total, 20 mg vascular tissue was cut into pieces and boiled for 10 min after adding l N acetic acid. Then, the vascular tissue was made into a homogenate in buffer (pH 7.4) containing 0.1 mol/L NaC1, 0.05 mol/L NaH2PO4, 0.05 mol/L Na2HPO4, 5 mmol/L EDTA and 5 mmol/L β-hydroxyquinoline. The plasma and vascular tissue supernatant was added to a Sep-Pak C18 column (Millipore – Waters, Milford, MA, USA) and eluted by the use of acetonitrile and trifluoroacetic acid and then redissolved with 100μL RIA buffer. The content of Ang II and Aldo was determined by the use of Ang II and Aldo RIA kits.
Semi-quantitative RT-PCR of OPN mRNA level in vascular tissue
Total aortic RNA was prepared with the use of TRIZOL reagent (Invitrogen, Carlsbad, CA, USA). In total, 1 μg RNA was reverse-transcribed into single-strand cDNA with the use of MMLV reverse transcriptase and oligo (dT) 15 primers. Primers for OPN were sense, 5′ CTC GCG GTG AAA GTG GCT GA 3′, and antisense, 3′ GAC CTC AGA AGA TGA ACT CT 5′; and for β-actin, sense, 5′ ATC TGG CAC CAC ACC TTC 3′, and antisense, 5′ AGC CAG GTC CAG ACG CA 3′. PCR was performed in a 0.2-mL tube containing 2μL tissue cDNA, 5μmol/L per each OPN sense and antisense primer mixture, 1 μL; 2.5 mmol/L per each dNTP mixture, 1 μL; 1.5 mmol/L MgCl2, 1.5 μL; 10× PCR buffer, 2.5 μL; and 1.25 unit Taq DNA polymerase, in a total volume of 25 μL. After being denatured at 95°C for 5 min, the solution underwent PCR at 94°C 30 s, 58°C 30 s and 72°C 40 s for 30 cycles, and then 72°C for 5 min. In total, 2-μL PCR product was separated in a 1.5% agarose gel and stained with ethidium bromide. The optical density of the 871-bp band was measured by the Gel Documentation System (Bio-Rad, Hercules, CA, USA). Amplification of OPN cDNA was confirmed by DNA sequencing. In total, 2-μL PCR product was amplified again with the β-actin primers at 94°C 30 s, 55°C 30 s and 72°C 40 s for 30 cycles, and then 72°C 5 min, and the optical density of the β-actin band (291 bp) was measured. The ratio of OPN to β-actin mRNA was considered the relative amount of OPN mRNA.
Statistical analysis
All data are expressed as mean ± SD. Student's t test was used to compare results for two groups and one-way analysis of variance, followed by a Student–Newman–Keuls test, to compare results for more than two groups. A P < 0.05 was considered statistically significant.
Results
VC induced by VDN in rats
Hemodynamic data and total vascular calcium content, alkaline phosphatase activity and 45Ca2+ uptake in rats
MAP, mean arterial pressure; LVESP, left ventricular end systolic pressure; LVEDP, left ventricular end diastolic pressure; ±LV dp/dt max, left ventricular peak rate of contraction and peak rate of relaxation; ALP, alkaline phosphatase; CPM, count per minute; VDN, rats given vitamin D3 plus nicotine for four weeks; VDN + captopril, vascular calcification, then captopril (angiotensin-converting enzyme inhibitor) administered by gavage for four weeks; VDN + spironolactone, vascular calcification, then spironolactone (Aldo receptor antagonist) administered by gavage for four weeks
Data are expressed as mean ± SD. n = 8
*P < 0.05, **P < 0.01 versus control; ## P < 0.01 versus VDN
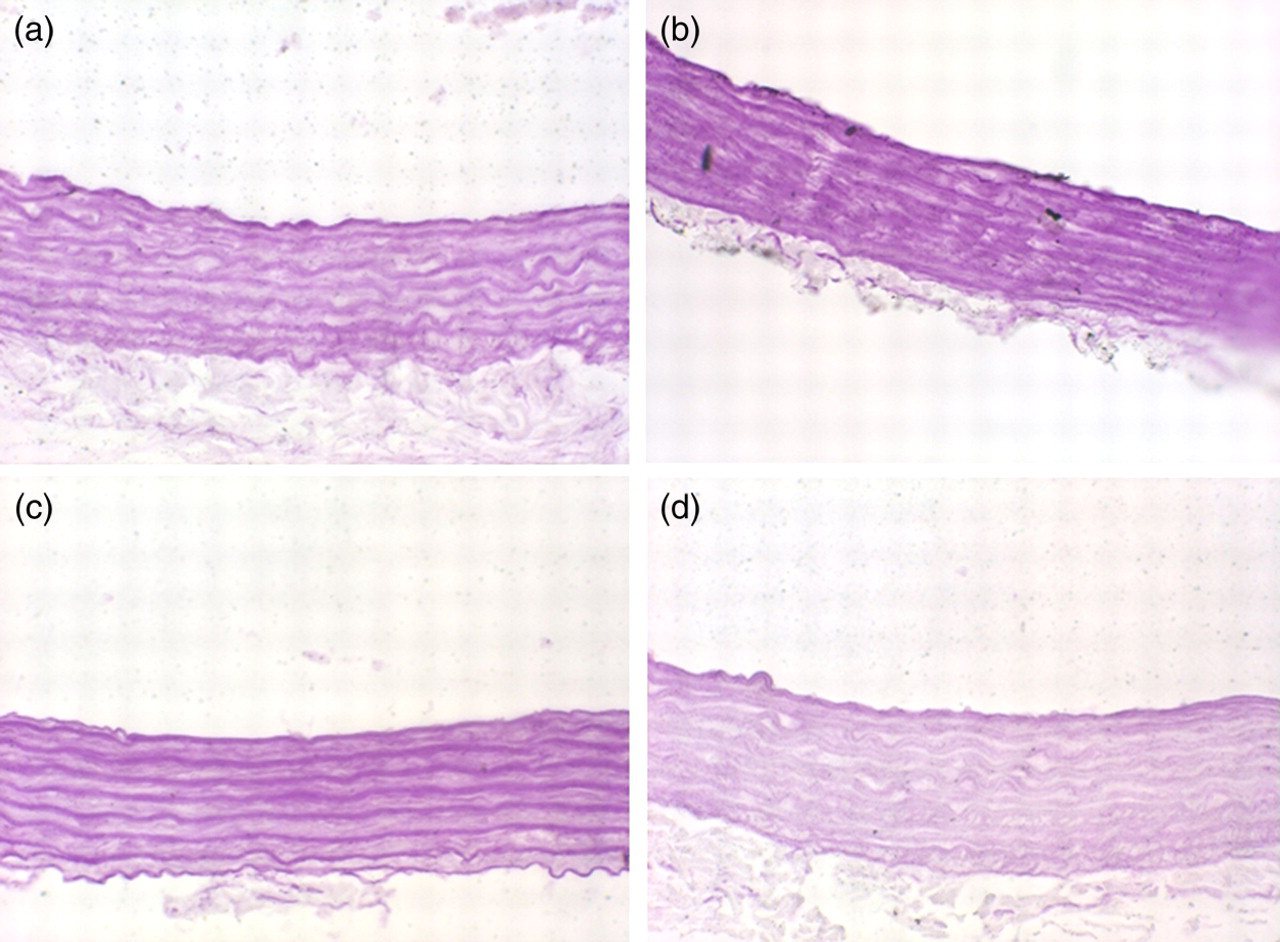
Von Kossa staining of calcification of multicellular nodules in elastic fibers of the medial layer in aortas (original magnification ×40). (a) Control; (b) VDN (rats given vitamin D3 plus nicotine for 4 weeks); (c) VDN + captopril (vascular calcification, then captopril [angiotensin-converting enzyme inhibitor] administered by gavage for 4 weeks); (d) VDN + spironolactone (vascular calcification, then spironolactone [Aldo receptor antagonist] administered by gavage for 4 weeks). (A color version of this figure is available in the online journal)

Sirius red staining of arteries for collagen deposition (original magnification ×40). (a) Control; (b) VDN (rats given vitamin D3 plus nicotine for 4 weeks); (c) VDN + captopril (vascular calcification, then captopril [angiotensin-converting enzyme inhibitor] administered by gavage for 4 weeks); (d) VDN + spironolactone (vascular calcification, then spironolactone [Aldo receptor antagonist] administered by gavage for 4 weeks). (A color version of this figure is available in the online journal)

Semi-quantitative reverse transcriptase polymerase chain reaction analysis of mRNA expression of osteopontin (OPN) in aortas of rats. **P < 0.01 versus control group; ## P < 0.01 versus VDN group. Data are mean ± SD from three experiments; n = 8. VDN, rats given vitamin D3 plus nicotine for four weeks; VDN + captopril, vascular calcification, then captopril (angiotensin-converting enzyme inhibitor) administered by gavage for four weeks; VDN + spironolactone, vascular calcification, then spironolactone (Aldo receptor antagonist) administered by gavage for four weeks
Content of Ang II and Aldo in plasma and vascular tissue with VC
Levels of angiotensin II and aldosterone in plasma and aortic tissue of rat without (control) or with induction of vascular calcification with vitamin D3 and nicotine (VDN)
Data are mean ± SD; n = 8; **P < 0.01 versus control
Cardiac function with VC was reduced and was improved with ACEI and Aldo receptor antagonist treatment
The mean arterial pressure and HR did not differ between rats with and without VC (P > 0.05), but the cardiac systolic and diastolic function was significantly reduced with calcification. LV dp/dt max and LV dp/dt min were 27% and 34% lower than that in the control group (P < 0.05 and P < 0.01), respectively. LVESP and LVEDP were higher by 42% and 32%, respectively, with VC than without VC (both P < 0.01, Table 1). As compared with VDN treatment alone, treatment with ACEI and Aldo receptor antagonist significantly improved cardiac function: LV dp/dt max increased by 11% and 24%, respectively, and LV dp/dtmin increased by 24% and 38%, respectively (all P < 0.05 and P < 0.01); LVESP was reduced by 36% and 29%, respectively, and LVEDP was decreased by 41% and 51% (all P < 0.01, Table 1).
ACEI and Aldo receptor antagonist treatment could reduce VC
Von Kossa staining showed that calcium deposition in vascular tissue significantly decreased with ACEI and Aldo receptor antagonist treatment (Figure 1). Sirius red staining showed lower vascular media fibrosis with ACEI than VDN alone, but no fibrosis was seen with Aldo receptor antagonism and there was no significant difference as compared with control treatment (Figure 2). With ACEI and Aldo receptor antagonist treatment, aortic calcium content, 45Ca2+ accumulation and ALP activity were 37%, 59% and 62% (all P < 0.01), and 32%, 44% and 60% (all P < 0.01), respectively, lower than with VDN treatment alone (Table 1). With ACEI and Aldo receptor antagonist treatment, vascular OPN mRNA level was 30% and 29% lower, respectively, than with VDN treatment alone (P < 0.01) (Figure 3).
Discussion
VC is a basic pathological process in atherosclerosis, hypertension, diabetes, chronic kidney disease, and aging. In calcified blood vessels, the decrease in vasodilator function and the increase in stiffness with VC easily lead to thrombosis and plaque rupture, which is an important risk factor of cardiovascular events. Aldo causes retention of sodium water and can promote collagen deposition and fibrosis, thus leading to fibrosis and structural remodeling of the heart. In this study, we used high doses of vitamin D3 to create VC in rats and found endogenous Aldo involved in VC. An increase in Ang II and Aldo levels in aorta was associated with increased calcium deposition in the arterial wall in rat aortas with VC
In this study, rats treated with high doses of vitamin D3 showed typical VC, which has several similarities to calcification in human athero- and arteriosclerosis: increased calcium content, as well as systolic hypertension and low body weight (data not shown), which agrees with results of several previous studies by our laboratory and others. 5,15,16 We found significantly increased content of Ang II and Aldo with VC and increased deposition of collagen. ACEI and Aldo receptor antagonist treatment reduced the overall Aldo production with VC, blocked the role of Aldo, significantly improved cardiac function, and ameliorated complications, especially the degree of VC and calcium deposition in the media and ALP activity. Furthermore, the agents reduced the mRNA expression of OPN, the organic component of bone, and tissue fibrosis. Therefore, Aldo takes part in the overall incidence of VC in rat and may be an endogenous bioactive factor in the pathogenesis of VC.
Aldo was previously thought to be produced only in adrenal glomerulosa cells, but an Aldo synthase CYP11B2 gene was found to be expressed by both vascular endothelial cells and smooth muscle cells, which suggests that extra-adrenal organs may also synthesize Aldo. 17,18 Various factors such as adrenocorticotropic hormone, Ang II, potassium, norepinephrine, endothelin and 5-hydroxytryptamine may stimulate the secretion of Aldo. 19–21 Aldo receptors exist in the kidney and in endothelial cells of large blood vessels. 22 Aldo can regulate the balance of water and electrolytes in the body and local tissue, but can also cause vascular fibrosis and endothelial dysfunction, directly damage vascular tissue and induce thrombosis. 23–25 Aldo promotes the proliferation of vascular smooth muscle cells and induces fibroblasts to synthesize and release collagen by interacting with MRs. During myocardial injury and heart failure, the level of Ang II and Aldo in heart and plasma is significantly increased and is positively correlated with severity of disease. 12 Coronary artery smooth muscle cells can produce Aldo and express MRs, and Aldo, by activating MRs, can promote the calcification of vascular smooth muscle cells in vitro. 14 In addition, Aldo can stimulate gene transcription of MRs at physiological hormone levels (1 nmol/L). 13 Aldo may mediate some Ang II-induced vascular effects in hypertension, in part by increased oxidative stress. 26 Aldo or Ang II may both regulate nuclear and activate gene transcription by MRs. The Aldo–MR complex enters the nucleus and acts as a nuclear transcription factor to bind to the MR response element, regulating the transcription of target genes such as collagen I and III, parathyroid hormone receptor 2, bone morphogenetic protein-2, ALP and interleukin-6, thus resulting in tissue fibrosis, VC and inflammation. 13 Our results confirm that Aldo takes part in VC.
Injection of Aldo in rats after a high-salt diet causes changes in metabolism, thus leading to hypocalcemia, hypomagnesemia, hyperparathyroidism, bone resorption and organizations, including calcium overload in the heart. 27 This is relevant to increasing the active oxygen and inflammation phenotype, which reduces cardiovascular function and promotes heart failure. Aldo plays a role in vascular smooth muscle and myocardia in heart and blood vessels and causes vascular injury by inducing nuclear swelling and stiffness in vascular endothelial cells. 28,29 In the heart and vascular tissue, Aldo causes membrane invagination and lipid deposition in part by upregulating the expression of endothelin-1 (ET-1) gene and in part by crosstalk with the ET-A receptor. Through the production of NADPH oxidase, xanthine oxidase and mitochondria free radicals increase free-radical activity. Increasing oxidative stress can activate inflammatory mediator nuclear factor-kappa B and activator protein 1, and increase adhesion molecules to cause vascular inflammation. 30 The Aldo receptor antagonist spironolactone can stabilize blood pressure and increase synthesis of NO synthase to improve endothelial function, inhibit the damage of Aldo to baroreceptors, and improve the baroreflex and compliance of the main artery. 31 Through MRs, Aldo can increase the mRNA and protein level of Ki-ras2A (a small GTP-binding protein) by a genome mechanism. In addition, Aldo promotes Na+/H+ exchange to strengthen vascular smooth muscle cells and increases the intracellular calcium and phosphorylation of extracellular signal-regulated kinase 1/2, thereby promoting calcium overload in cells and their proliferation. 32 This effect can be blocked by the Aldo receptor antagonist spironolactone and by nucleic acid and protease inhibitors. We found the local secretion of Aldo increased in rats with VC. With spironolactone treatment, VC was reduced, so Aldo may affect the incidence of VC, but its precise mechanism needs further study.
In the present study, we found the tissue level of Aldo increased with VC induction in rats. ACEI and Aldo receptor antagonist treatment could significantly improve cardiac function with VC, and the degree of VC was reduced, which suggests that Aldo is involved in cardiac VC as a paracrine/autocrine factor. We provide a new understanding of the role of Aldo in cardiovascular diseases. Aldo receptor antagonism may be effective in preventing and treating cardiovascular diseases. However, the interaction between Aldo and other regulatory factors such as Ang II, oxidative stress and cytokines, as well as the signal transduction and molecular mechanism, need further study.
Footnotes
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (grant nos. 30770869 and 30871013 to Y-FQ, and 81070090 to C-SX) and a co-construction project of Beijing Municipal Commission of Education.