
Abstract
The present study focused on the isolation, cultivation and characterization of human mesenchymal stem cells (MSCs) from adipose tissue and on their differentiation into chondrocytes through the NH ChondroDiff medium. The main aim was to investigate some markers of biomechanical quality of cartilage, such as lubricin, and collagen type I and II. Little is known, in fact, about the ability of chondrocytes from human MSCs of adipose tissue to generate lubricin in three-dimensional (3D) culture. Lubricin, a 227.5-kDa mucinous glycoprotein, is known to play an important role in articular joint physiology, and the loss of accumulation of lubricin is thought to play a role in the pathology of osteoarthritis. Adipose tissue is an alternative source for the isolation of multipotent MSCs, which allows them to be obtained by a less invasive method and in larger quantities than from other sources. These cells can be isolated from cosmetic liposuctions in large numbers and easily grown under standard tissue culture conditions. 3D chondrocytes were assessed by histology (hematoxylin and eosin) and histochemistry (Alcian blue and Safranin-O/fast green staining). Collagen type I, II and lubricin expression was determined through immunohistochemistry and Western blot. The results showed that, compared with control cartilage and monolayer chondrocytes showing just collagen type I, chondrocytes from MSCs (CD44-, CD90- and CD105- positive; CD45-, CD14- and CD34-negative) of adipose tissue grown in nodules were able to express lubricin, and collagen type I and II, indicative of hyaline cartilage formation. Based on the function of lubricin in the joint cavity and disease and as a potential therapeutic agent, our results suggest that MSCs from adipose tissue are a promising cell source for tissue engineering of cartilage. Our results suggest that chondrocyte nodules producing lubricin could be a novel biotherapeutic approach for the treatment of cartilage abnormalities.
Keywords
Introduction
Lubricin, a 227.5-kDa mucinous glycoprotein, is a proteoglycan synthesized and expressed by articular chondrocytes of the superficial zone, 1 which plays an important role in articular joint physiology. The loss of accumulation of lubricin may play a role in the pathology of osteoarthritis (OA). Lubricin interacts with the articular surface and functions as a boundary lubrificant in articular cartilage joints and reduces the coefficient of friction of the articular cartilage surface. 2 The content of this protein is damaged with aging and OA, and the lubricin gene is differently expressed in the synovium of rheumatoid arthritis (RA) and OA, implying a possible role in the pathogenesis of these diseases. 3 Recently, it was suggested that recombinant lubricin molecules might be used in novel biotherapeutic approaches for the treatment of OA and associated cartilage abnormalities. 4
Regenerative medicine is an emerging field that seeks to repair or replace injured tissues and organs through natural or bioengineered means. Autologous chondrocyte implantation is an accepted surgical treatment in patients with isolated cartilage defects, but there are several limiting factors when using autologous chondrocytes. Each biopsy is an additional trauma to cartilage that is already damaged and injured in the joint. Furthermore, suitable donor sites for hyaline cartilage are rare in damaged joints, and as yet, other cartilage has not been fully assessed for its ability to repair articular cartilage. 5 In addition, the number of cells obtained via biopsy is relatively small and therefore the cells have to be expanded for several passages. Chondrocytes grown in monolayer de-differentiate and subsequently lose their chondrogenic phenotype and their re-differentiation potential. This loss of cartilage-specific matrix components is indicated mainly through a switch in the cells from producing collagen type II to collagen type I. 6,7 It has been shown that re-differentiation is possible with the help of specific growth factors such as transforming growth factor-β as well as without growth factors. 8–10 However, to avoid further distress for the traumatized joint and initially obtain high numbers of cells, an alternative source of cells with chondroprogenitor potential would be useful. One such possibility for the construction of neo-cartilaginous tissue is the use of mesenchymal stem cells (MSCs). 11
MSCs have generated a great deal of public, scientific and media interest because of their potential use in regenerative medicine and tissue engineering. Stem cells have the remarkable potential to develop into many different cell types in the body. Recent research on stromal MSCs has provided a new and exciting opportunity for bone and cartilage tissue engineering. Thus far, MSCs have been isolated from bone marrow, periosteum, trabecular bone, adipose tissue, synovium, skeletal muscle and deciduous teeth. 12 MSCs possess the capacity to differentiate into cells of connective tissue lineages, including bone, 13–15 fat, 12,16 cartilage, 12,13 intervertebral disc, 14 ligament 17,18 and muscle. 12,19
Adipose tissue is an alternative stem cell source that can be obtained by less invasive methods and in larger quantities than bone marrow. It has been demonstrated that adipose tissue contains stem cells similar to bone marrow MSCs, which are termed processed lipoaspirate (PLA) cells. 20 These cells can be isolated from cosmetic liposuctions in large numbers and grown easily under standard tissue culture conditions. The multilineage differentiation capacity of PLA cells has been confirmed. It has also been observed that human adipose tissue yields higher amounts of MSCs than bone marrow or umbilical cord blood. 21,22
The International Society for Cellular Therapy has listed the main factors required for a cell to be regarded as an MSC in a position statement. 23 MSCs are characterized through their adhesion potential in monolayer culture and through their differentiation potential into chondrocytes, osteocytes and adipocytes in vitro. Furthermore, the International Society for Cellular Therapy has listed several markers that cells should exhibit or lack in order to be classified as MSCs. MSC positive markers include CD44, CD90 and CD105, whereas MSCs should lack CD45, CD14 and CD34. 23
This study focused on the isolation, cultivation and characterization of human MSCs from adipose tissue and on their differentiation through a three-dimensional (3D) environment to stimulate chondrogenic signalling pathways in the MSCs. This medium promotes the differentiation and further maturation of marrow stromal cells from adipose tissue into chondrocytes. Differentiated chondrocytes in micromass culture form 3D clusters termed chondrocyte ‘nodules’. In this research, we evaluated matrix production through histology using hematoxylin and eosin staining, and synthesis of extracellular matrix (ECM) molecules, such as glycosaminoglycan (GAG), by more specific chemical stains such as Safranin-O and Alcian blue. Immunohistochemistry and Western blot were used to detect and quantify type I (fibro-cartilage), type II (hyaline cartilage) collagens and lubricin. The latter provides essential chondroprotective properties to articular cartilage as evidenced by lack of cartilage surface integrity and surface disruption in lubricin knockout mice. 24 The in vitro expansion and differentiation of MSCs to chondrocytes producing lubricin for tissue repair might be an important first step for the future treatment of joint diseases, 25 including osteochondrodysplasias, non-union fractures and OA. MSCs from adipose tissue exhibiting chondroprotective lubricin may represent a useful cell source, as an alternative approach to the treatment of cartilage defects, due to their availability in relatively high numbers and the ease of obtaining them.
Material and methods
Patients
Adipose tissue was gathered from ten donors, five men and five women (from 22 to 30 years of age) undergoing liposuction procedures. Lipoaspirates were obtained under an approved Institutional Review Board protocol and after informed consent had been obtained from the patients at the Cannizzaro Hospital, Catania (Italy). The patients were not smokers and occasionally taking non-steroidal anti-inflammatory drugs (NSAIDs). The women did not take estrogen replacement therapy.
Cultures of human MSCs from adipose tissue
The raw lipoaspirate (50–100 mL) was washed with sterile phosphate-buffered saline (PBS; Invitrogen, Milan, Italy) to remove red blood cells and debris, and incubated for three hours at 37°C with an equal volume of serum-free Dulbecco's modified Eagle's medium (DMEM)-low glucose (DMEM-1g; PAA Laboratories, Pasching, Austria) containing 0.075% of type I collagenase (Invitrogen). Collagenase activity was then inactivated by an equal volume of DMEM-1g containing 10% of heat-inactivated fetal bovine serum (FBS; Invitrogen). Successively, the digested lipoaspirate was centrifuged at 1200 rpm for 10 min. The pellets were re-suspended in PBS (plus penicillin/streptomycin 1%) and filtered through a 100-μm nylon cell strainer (Falcon BD Biosciences, Milan, Italy). The filtered cells were again centrifuged at 1200 rpm for 10 min, plated in T75 culture flasks (Falcon BD Biosciences) with DMEM-1g (10% FBS, penicillin/streptomycin 1%) containing 1% of MSC growth medium (MSCGS; ScienCell Research Laboratories, Milan, Italy) and incubated at 37°C with 5% CO2. Twenty-four hours after the initial plating, no adherent cells were removed by intensely washing the plates.
Differentiation of human MSCs in chondrocytes
MSCs were trypsinized and diluted to a final concentration of 2.5 × 105 cells/mL medium. Then, 1 mL of cell suspension was transferred in a 15-mL polypropylene conical tube. They were centrifuged at 700 rpm for five minutes, and after removing the medium, 1 mL of NH ChondroDiff medium (MACS; Miltenyi Biotec, Bologna, Italy) containing penicillin/streptomycin 1% was added. The pellet was re-suspended carefully and centrifuged at 700 rpm for further five minutes to form a pellet. With no further re-suspension, they were cultured for 24 days at 37°C and 5% CO2 in 1 mL of NH ChondroDiff medium, changed every third day.
Human chondrocyte isolation and monolayer culture
Normal human articular cartilage was obtained at replacement surgery from some patients with femoral neck accidental fractures from whom informed consent was obtained. The isolation procedure was conducted under antiseptic conditions. The cartilage was cut into small fragments and carefully washed using DMEM culture medium containing NaHCO3, 25 mmol/L HEPES, 1 mmol/L sodium pyruvate, 50 μg/mL gentamycin, 100 U/mL penicillin, 100 μg/mL streptomycin and 2.5 μg/mL amphotericin B. Chondrocytes were isolated through three sequential passages of enzymatic digestion of the ECM: incubation with 0.1% hyaluronidase type III (1 mg/mL for 100 mg of cartilage), for 30 min at 37°C; incubation with 0.5% pronase type XIV (5 mg/mL for 100 mg of cartilage), for 60 min at 37°C; finally incubation with 0.2% collagenase type IA (2 mg/mL for 100 mg of cartilage), for 45 min at 37°C. The obtained cellular suspension was filtered (filters from 100 and 70 μm) to eliminate the residues of the digestion, cellular aggregates and to obtain a monocellular suspension of chondrocytes. This was washed three times with DMEM supplemented with 10% fetal calf serum, and was subjected to vital coloration method staining with trypan blue in order to determine the number and the vitality of recovered cells. Monolayer chondrocytes were plated in 100-mm Petri dishes (for Western blot) and cells maintained for 24 d. The medium was changed every 2–3 d.
Determination of MSC markers
After reaching confluence (80% of total flask surface), all subpopulations were trypsinized (Sigma-Aldrich, Milan, Italy) and subcultured in a 12-well culture dish for two days. Immunocytochemistry was carried out on MSCs to identify the specific markers that characterize this type of cells. Cells were first washed with PBS, then fixed with 4% paraformaldheyde in PBS for 30 min and incubated for 30 min with a 5% solution of normal goat serum (Sigma-Aldrich). They were subsequently incubated overnight at 4°C with primary antibodies (Millipore, Milan, Italy): CD44, 1:200 dilution; CD90, 1:100; CD105, 1:100; CD14, 1:200; CD34, 1:200; CD45, 1:200. The following day, cells were washed with PBS and incubated for one hour at room temperature with Cy3-conjugated goat anti-rabbit secondary antibody or Cy3-conjugated goat anti-mouse secondary antibody or fluorescein isothiocyanate-conjugated goat anti-mouse secondary antibody (Millipore). As a control, the specificity of immunostaining was verified by omitting incubation with the primary or secondary antibody. Digital images were acquired using a Leica DMRB fluorescence microscope (Leica Microsystems Srl, Milan, Italy) equipped with a computer-assisted Nikon digital camera (Nital SpA, Turin, Italy). Immunoreactivity was evaluated taking into account the signal-to-noise ratio of immunofluorescence.
Histology and histochemistry
Twenty-four days from the start of MSC differentiation, the chondrocyte nodules were rinsed in PBS, and fixed in 4% paraformaldehyde. After an overnight wash, specimens were dehydrated in graded ethanol, cleared in xylene and paraffin-embedded, preserving their anatomical orientation. Sections, 4–5-μm thick, were cut from paraffin blocks using a microtome and they were mounted on sialane-coated slides and stored at room temperature. The sections were stained with hematoxylin and eosin (H&E) for general cell identification and Alcian blue and Safranin-O/fast green staining for proteoglycan detection according to the protocol of the manufacturer (Bio-Optica, Milan, Italy). Sections were observed with an Axioplan Zeiss light microscope (Carl Zeiss, Oberkochen, Germany) and photographed with a digital camera (Canon, Tokyo, Japan).
Immunohistochemistry
For immunohistochemical analysis, sections were incubated for 30 min in 0.3% H2O2/methanol to quench endogenous peroxidase activity, and then rinsed for 20 min with PBS (Bio-Optica). The sections were irradiated (5 min × 3) in capped polypropylene slide-holders with citrate buffer (pH 6), using a microwave oven (750 W) to unmask antigenic sites. Then, the sections were incubated with diluted rabbit polyclonal antibody against collagen type I and II (Research Diagnostics, Inc, Flanders, NJ, USA), 1:100 working dilution, respectively, and lubricin (Novus Biologicals, LLC, Littleton, CO, USA), 1:50 working dilution, overnight at 4°C. After, the secondary antibody, biotinylated mouse/anti-rabbit IgG was applied (for 30 min, at room temperature), followed by the avidin–biotin–peroxidase complex (Vector Elite Kit; Abbott, Chicago, IL, USA) for 30 min, at room temperature. The immunoreactions were visualized by incubating the sections for four minutes in a 0.1% 3,3′-diaminobenzidine and 0.02% hydrogen peroxide solution (DAB substrate kit; Vector Laboratories, Milan, Italy). The sections were lightly counterstained with Mayer's hematoxylin (Histolab Products AB, Goteborg, Sweden), mounted in GVA mount (Zymed Laboratories Inc, San Francisco, CA, USA), observed with an Axioplan Zeiss light microscope (Carl Zeiss) and photographed with a digital camera (Canon).
Evaluation of immunohistochemistry: the collagen type I, II and lubricin-staining status was identified as either negative or positive. Positive staining was defined as the presence of a brown detection chromogen on the edge of the hematoxylin-stained cell nucleus, distributed within the cytoplasm or in the immediate lacunar/pericellular space. Stain intensity and the proportion of immunopositive cells were assessed also by light microscopy. Intensity of staining (IS) was graded on a scale of 0–4, according to the following assessment: 0 = no detectable staining; 1 = weak staining; 2 = moderate staining; 3 = strong staining; and 4 = very strong staining. The percentage of collagen type I, II and lubricin immunopositive cells (ES, Extent Score) was independently evaluated by two investigators (one anatomical morphologist and one histologist) and scored as a percentage of the final number of 100 cells in five categories: 0−5%; + = 5–30%; ++ = 31–50%; ++ + =51–75% and ++ + + >75%. Counting was performed at ×200 magnification.
Positive and negative controls were performed to test the specific reaction of primary antibodies used in this study at a protein level. For positive control testing, the sections from lung cartilage underwent an immunoperoxidase process. The positive immunolabeling for collagen type I, II and lubricin were perinuclear and cytoplasmic. Negative controls were incubated without the primary antibody.
Western blot analysis
The expression of collagen type I, type II and lubricin was evaluated by Western blot analysis. Briefly, the monolayer and chondrocyte nodules were washed twice with ice-cold PBS and collected with lysing buffer (10 mmol/L Tris-HCl plus 10 mmol/L KCl, 2 mmol/L MgCl2, 0.6 mmol/L PMSF and 1% SDS, pH 7.4). After cooling for 30 min at 0°C, cells were sonicated. Twenty micrograms of total protein, present in the supernatant, were loaded on each lane and separated by 4–12% Novex Bis-Tris gel electrophoresis (NuPAGE; Invitrogen). Proteins were then transferred onto nitrocellulose membranes (Invitrogen) in a wet system. The transfer of proteins was verified by staining the nitrocellulose membranes with Ponceau S and the Novex Bis-Tris gel with Brillant blue R. Membranes were blocked in Tris-buffered saline containing 0.01% Tween-20 (TBST) and 5% non-fat dry milk at 4°C overnight. Rabbit polyclonal anticollagen types I and II (1:100 dilution; Research Diagnostic, Inc), lubricin (1:50 dilution), and mouse monoclonal anti-α-tubulin antibodies (Sigma, Milan, Italy; 1:5000) were diluted in TBST and membranes were incubated for 24 h at room temperature. Antibodies were detected with horseradish peroxidase-conjugated secondary antibody using the enhanced chemiluminescence detection Supersignal West Pico Chemiluminescent Substrate (Pierce Chemical Co, Rockford, IL, USA). Bands were measured densitometrically and their relative density was calculated based on the density of the α-tubulin bands in each sample. Values were expressed as arbitrary densitometric units (ADU) corresponding to signal intensity.
Statistics
All data were normally distributed. Comparisons between two means were tested with the Student's t-test; P values of less than 0.05 were considered statistically significant. Ten fields from randomly selected slides were observed under a light microscope. Each field was photographed with a digital camera. On each photomicrograph, three observers, blinded to the type of sample, identified and counted the number of total cells as well as the number of these cells exhibiting a positive reaction. The proportion of positive cells was calculated for each photomicrograph and a mean value was obtained for each sample. The results were expressed as a percentage. All data were analyzed with the SPSS program (SPSS® release 16.0, Chicago, IL, USA). Cohen's kappa was applied to measure the agreement between the three observers and averaged over all three to evaluate overall agreement using the following grading: 0–0.2 (slight); 0.21–0.40 (fair); 0.41–0.60 (moderate); and 0.61–0.80 (substantial); and 0.81–1.0 (almost perfect).
Results
Characterization of MSCs
In order to identify MSCs derived from lipoaspirate, immunocytochemical procedures were carried out using several cell surface markers. In particular, they were recognized for their immunoreactivity to MSC-specific cell type markers, rather than hematopoietic stem cell markers. As shown in Figure 1, MSCs were marked by CD44 (A), CD90 (B) and CD105 (C), but were not immunopositive for CD45 (D), CD14 (E) and CD34 (F).

CD44 (a), CD90 (b), CD105 (c) (positive markers), CD45 (d), CD14 (e) and CD34 (f) (negative markers) mesenchymal stem cell (MSC) marker expression by immunocytochemistry and using a Leica DMRB fluorescence microscope equipped with a computer-assisted Nikon digital camera (a–f). Magnification ×40; scale bar: 50 μm. (A color version of this figure is available in the online journal)
Histology and histochemistry
The results of histology (H&E staining) and histochemistry (Alcian blue and Safranin-O/fast green staining) demonstrated the absence of structural alterations.
The H&E staining was used to determine the cell vitality in differentiated chondrocytes in 3D cultures (Figures 2a–c). The chondrocytes showed an increase in proliferation, ECM production and cellular aggregation (a like-typical structure of the hyaline cartilage). They did not show any signs of cellular suffering demonstrated by an intense (++ + , strong) staining.
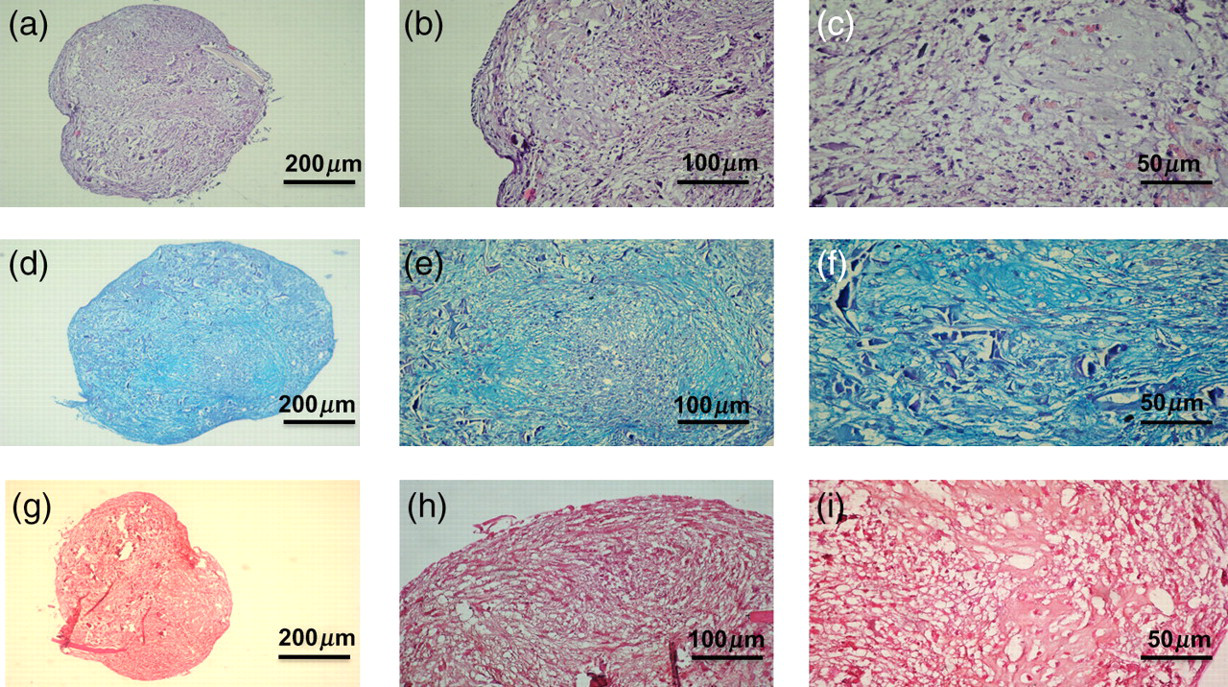
Hematoxylin and eosin (a–c), Alcian blue (d, e) and Safranin-O/fast green (g–i) staining of chondrocytes in micromass culture forming three-dimensional clusters termed chondrocyte ‘nodules’. (a–c) Intense (strong) staining demonstrates increase in proliferation, cellular aggregations (like-typical structures of the hyaline cartilage), extracellular matrix production and no sign of cellular suffering. (d,e) An intense (strong) Alcian blue staining demonstrates the presence of proteoglycans. (g–i) Intense (strong) Safranin-O/fast green staining demonstrates the presence of proteoglycans. (a) Magnification ×10, scale bar: 200 μm; (b) magnification ×20, scale bar: 100 μm; (c) magnification ×40, scale bar: 50 μm; (d) magnification ×10, scale bar: 200 μm; (e) magnification ×20, scale bar: 100 μm; (f) magnification ×40, scale bar: 50 μm; (g) magnification ×10, scale bar: 200 μm; (h) magnification ×20, scale bar: 100 μm; (i) magnification ×40, scale bar: 50 μm. (A color version of this figure is available in the online journal)
Specific chemical stains, such as Alcian blue and Safranin-O/fast green, were used to assess synthesis of GAG-containing proteoglycans (assessment on the IS). Differentiated chondrocytes showed the presence of proteoglycans demonstrating an intense (++ + , strong) Alcian blue (Figures 2d–f) and Safaranin-O/fast green (Figures 2g–i) staining.
Immunohistochemistry
Collagen type I was appreciated both in chondrocyte nodules and in the ECM. Expression of collagen type I is indicative of fibrocartilage formation. A moderate collagen type I staining was observed where many chondrocytes were immunolabeled (++) (Figures 3a and c). No immunoreaction was observed in the negative control treated with PBS without the primary antibody (Figures 3b and d). Both ES and IS were significantly greater (P < 0.01). Inter-observer agreement, measured using the Kappa coefficient, was 0.90 (almost perfect).
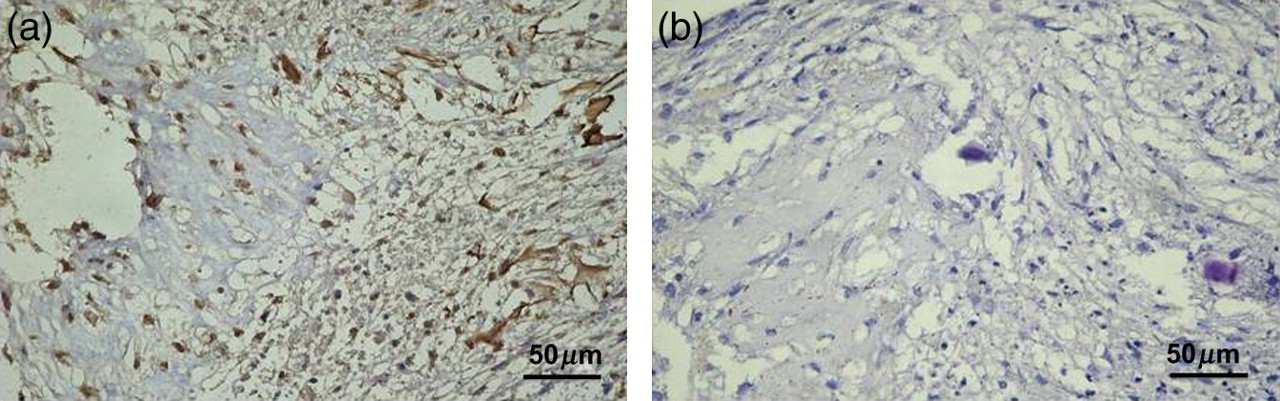
Collagen type I (a,b) immunohistochemistry specimen from chondrocytes in micromass culture forming three-dimensional clusters termed chondrocyte ‘nodules’. Expression of collagen type I is indicative of fibrocartilage formation. (a) A moderate collagen type I staining was observed where many chondrocytes were immunolabeled (++). (b) Negative control treated with phosphate-buffered saline without the primary antibodies. (a, b) Magnification ×40; scale bar: 50 μm. (A color version of this figure is available in the online journal)
Collagen type II was detected in cells and the ECM. Expression of collagen type II, indicative of hyaline cartilage formation, was investigated via immunohistochemistry in chondrocyte nodules. A strong collagen type II staining was observed where almost all chondrocytes were immunolabeled (+++) (Figures 4a and c). No immunoreaction was observed in the negative control treated with PBS without the primary antibodies (Figures 4b and d). Both ES and IS were significantly greater (P < 0.01). Inter-observer agreement, measured using the Kappa coefficient, was 0.92 (almost perfect).

Collagen type II (a,b) immunohistochemistry specimen from chondrocytes in micromass culture forming three-dimensional clusters termed chondrocyte ‘nodules’. Expression of collagen type II is indicative of hyaline cartilage formation. (a) A strong collagen type II staining was observed where almost all chondrocytes were immunolabeled (+++). (b) Negative control treated with phosphate-buffered saline without the primary antibodies. (a, b) Magnification ×40; scale bar: 50 μm. (A color version of this figure is available in the online journal)
Lubricin immunohistochemical staining in 3D cultures was appreciated both in cells and in the ECM. A very strong lubricin staining (grade 4) was observed where almost all chondrocytes were immunolabeled (++++) (Figures 5a and c). No immunoreaction was observed in the negative control treated with PBS without the primary antibodies (Figures 5b and d). Both ES and IS were significantly greater (P < 0.01). Inter-observer agreement, measured using the Kappa coefficient, was 0.94 (almost perfect).
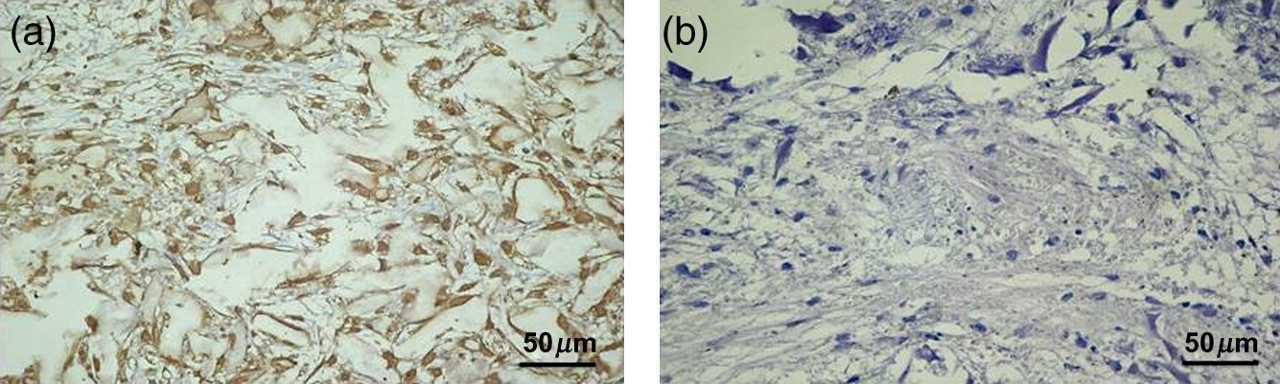
Lubricin (a,b) immunohistochemistry specimen from chondrocytes in micromass culture forming three-dimensional clusters termed chondrocyte ‘nodules’. (a) A very strong lubricin staining was observed where almost all chondrocytes were immunolabeled (++++). (b) Negative control treated with phosphate-buffered saline without the primary antibodies. (a,b) Magnification ×40; scale bar: 50 μm. (A color version of this figure is available in the online journal)
Western blot
In this study, we also examined, by Western blot, the production of collagen type I, type II and lubricin in the human monolayer and differentiated from MSC chondrocytes. The results confirmed the same behavior as immunohistochemistry. As shown in Figure 6, both human monolayer and chondrocytes differentiated from MSCs potently produced collagen type I. At 24 d, monolayer chondrocytes expressed collagen type II in a small quantity; whereas at the same time, chondrocytes differentiated from MSCs induced a strong expression of collagen type II (Figure 7). Moreover, we have investigated the effects of different cultures on the release of lubricin. Negligible amounts of lubricin were produced by monolayer chondrocytes. In contrast, lubricin was potently induced by chondrocytes differentiated from MSCs in 3D cultures (Figure 8).

Collagen type I expression induced in human monolayer chondrocytes (monolayer chondrocytes) and differentiated mesenchymal stem cell (MSC) chondrocytes (chondrocytes) determined by Western blot analysis. Data show the relative expression (mean ± SEM) of collagen type I calculated as arbitrary densitometric units (ADU) collected from three independent experiments
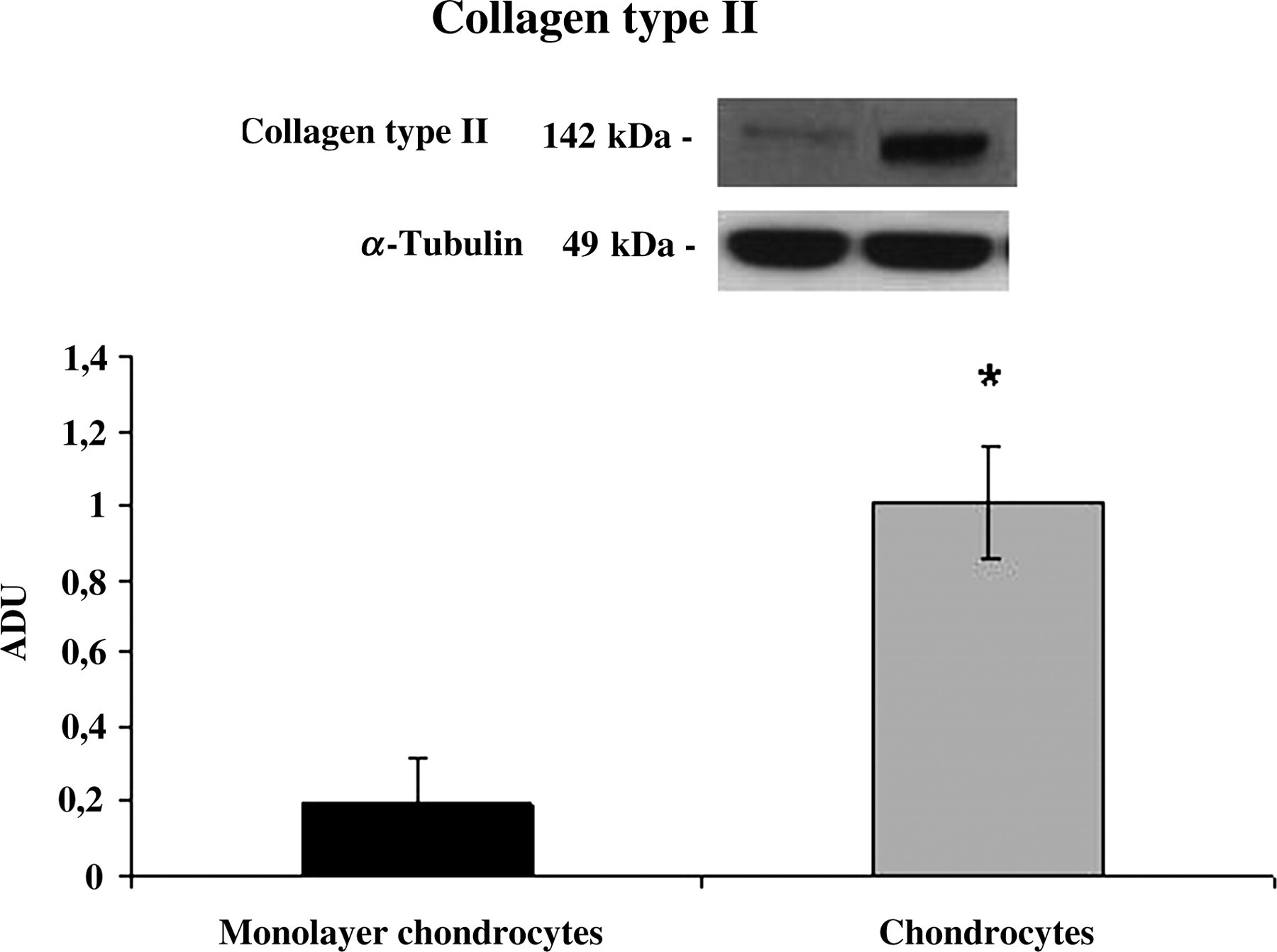
Collagen type II expression induced in human monolayer chondrocytes (monolayer chondrocytes) and differentiated from mesenchymal stem cell (MSC) chondrocytes (chondrocytes) determined by Western blot analysis. Data show the relative expression (mean ± SEM) of collagen type II calculated as arbitrary densitometric units (ADU) collected from three independent experiments. *P < 0.05 compared with monolayer chondrocytes
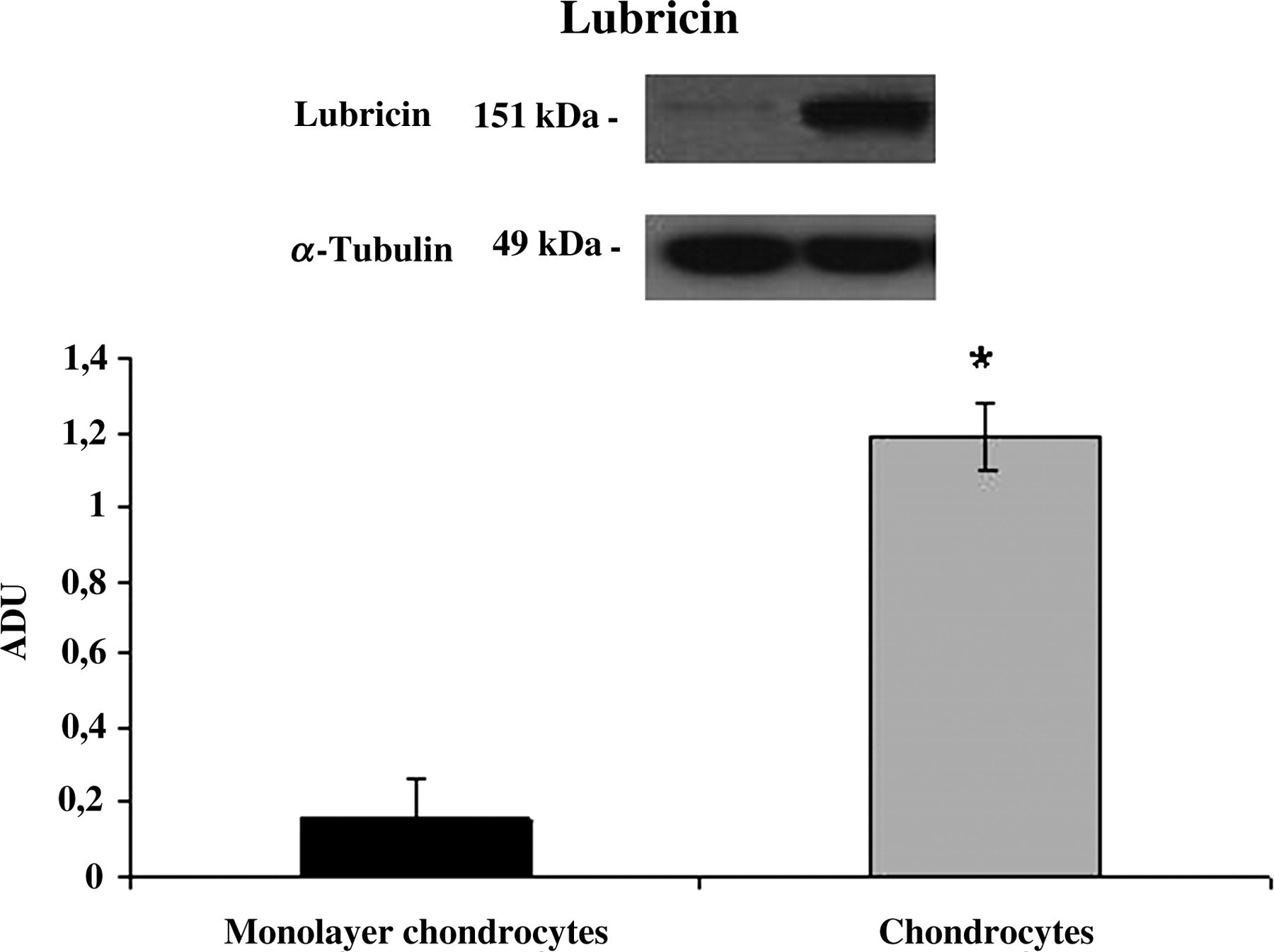
Lubricin expression induced in human monolayer chondrocytes (monolayer chondrocytes) and differentiated mesenchymal stem cell (MSC) chondrocytes (chondrocytes) determined by Western blot analysis. Data show the relative expression (mean ± SEM) of lubricin calculated as arbitrary densitometric units (ADU) collected from three independent experiments. *P < 0.05 compared with monolayer chondrocytes
Discussion
In this study, we have evaluated collagen type I, II and lubricin expression on paraffin sections, through histology, histochemistry, immunohistochemistry and Western blot, in chondrocytes differentiated from MSCs of adipose tissue. In our culture conditions, the chondrocytes formed 3D clusters termed chondrocyte nodules.
The results clearly demonstrated that MSCs from adipose tissue grown in 3D clusters were able to express collagen type I, II and lubricin. Compared with monolayer chondrocytes, which showed only collagen type I expression, characteristic of fibrous cartilage, the increased expression of collagen type II and lubricin (which are components of joint lubrication) is indicative of hyaline cartilage formation. This is also supported by the presence of proteoglycans.
Lubricin is a chondroprotective glycoprotein which acts as a vital counteragent against aberrant protein and/or cellular adhesion, infiltration and over-proliferation, and serves as a critical boundary lubricant between opposing cartilage surfaces. This protein is less expressed with aging and during OA; moreover, the lubricin gene is differently expressed in the synovium of RA and OA, implying a possible role in the pathogenesis of these disease. 2 It has been identified as a component of joint lubrication and synovial homeostasis by reducing fiction in joints and preventing synovial cell overgrowth. It is a large, water-soluble molecule that acts as a carrier for insoluble surface-active phospholipids. Depletion of lubricin function has been associated with camptodactyly-arthropathy-coxa vara-pericarditis syndrome and arthritic-like autosomal recessive disorder. Several recent studies found that treatment with recombinant lubricin could protect articular cartilage and prevent the process of OA in an animal model. 26 The synthesis and localization of lubricin have been found down-regulated in rat, sheep and guinea-pig models of OA, 27,28 which further suggest that augmentation of lubricin levels in OA joints could be a potential therapeutic for OA. Additionally, the stimulation of lubricin expression in chondrocytes near the articular surface may be useful for creating tissue-engineered cartilage from isolated subpopulations with a surface that is bioactive and functional in lubrication. 29 In fact, a recent study by Flannery and colleagues 4 demonstrated that intra-articular treatment with recombinant lubricin could prevent the degeneration of cartilage in a rat OA model. The 3D chondrocyte nodules have been demonstrated to produce lubricin, which could protect articular cartilage and prevent the process of OA. These findings, according to our knowledge, have not yet been demonstrated by anyone in the literature.
Cartilage is a connective tissue that owes its special characteristics to the dense ECM produced by chondrocytes. About 40–50% of cartilage ECM consists of collagens (of these, approximately 90% is collagen type II) and about 20–25% of different proteoglycans (aggrecan, decorin, biglycan and fibromodulin). A particular feature of its cells, the chondrocytes (which make up less than 5% of the tissue's total 3D volume), is that they do not have any direct cell-to-cell contact with each other. Thus, each cell may be regarded as a functional unit responsible for maintaining the ECM in its immediate surrounding through balanced and tightly regulated anabolic and catabolic activities. Cartilage is avascular, aneural and alymphatic. 30 This unique composition further explains the limited repair capacity of articular cartilage and why most repair tissues fail due to the dominance of a fibroblast-like cell type which produces an ECM without the necessary biomechanical properties of hyaline cartilage. 31,32 During embryogenesis, cartilage is formed from the condensation of MSCs. Mesenchymal cell aggregates, termed blastema in vivo, lead cartilage differentiation in vivo and also in high-density cell cultures. 33,34 This process is characterized by the production of cartilage matrix-specific proteoglycans and a switch from collagen type I to collagen type II synthesis. 35 Articular cartilage homeostasis is the result of an intricate interplay between anabolic and catabolic, anti- and proinflammatory, and anti- and proapoptotic mediators. These processes act also as protective mechanisms after sublethal injury characterized by an increased survival of chondrocytes, which play a central role in the growth of new cartilage and ECM. 36,37
Over the last decades, surgeons and researchers have been working hard to elaborate surgical cartilage repair interventions and to restore the surface of an articular joint's hyaline cartilage. In the case of cartilage tissue engineering, after expansion in monolayer culture, chondrocytes are seeded onto a 3D scaffold before transplantation into the defect. The properties of these scaffolds follow basic principles; they must be biocompatible, structurally and mechanically stable, and must support the loading of an appropriate cell source to allow successful infiltration and attachment to the host tissue. 38 The simplest approach of cartilage repair using MSCs can be done in a manner analogous to the autologous cell transplantation (ACT) method. After expanding the MSCs in vitro, they are injected into a cartilage defect. MSCs are hereby frequently combined with a soluble scaffold to ensure they remain in the cartilage defect similar to the fibrin glue or the periosteal flap used in classic ACT. The results have been ambiguous. Some have reported the formation of new cartilage, whereas others have shown degradation and fragmentation of the MSCs. Most studies are still in the experimental trial phase and have been performed using animal models, such as rabbit 39 and goat. 40,41 The ultimate goal of clinicians and scientists involved in cartilage and OA research is achieving better articular cartilage repair and eliminating or significantly reducing pain and inflammation while restoring a mechanically functional repair tissue.
The possibility of applying autologous cell transplantation in conjunction with chondrocyte nodules from adipose tissue producing lubricin for repairing cartilage lesions in patients with OA could be of great interest. Lubricin could reduce the progression of the articular diseases and prevent the process of OA.
We believe that the findings presented in this manuscript, though still preliminary data, could hold important clinical relevance, and have implications for medical treatment. Further studies are necessary to gain new insights into the behaviour of 3D chondrocyte nodules in in vitro systems and in animal models.
Conclusions
In conclusion, the present experiments establish that MSCs are a promising cell source for tissue engineering of articular cartilage. We demonstrate that adipose-tissue-derived human MSCs may be differentiated into chondrocytes in 3D culture. The resultant chondrocyte nodules may be promising therapeutics for the treatment of articular diseases, in particular in preventing the development and progression of OA and other cartilage degenerative pathologies.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Professor Maria Luisa Carnazza and Professor Giuseppa Martinez (Department of Bio-medical Sciences, University of Catania, Italy) for their comments and suggestions to the paper, and Professor Rosario Perrotta and Dr Maria Stella Tarico (Department of Plastic Surgery, University of Catania, Italy) for providing surgical material. This study was supported by grants provided by the Medicine and Surgery Faculty of University of Catania.