
Abstract
Helicobacter pylori (H. pylori) dysregulates the expression of various genes resulting in gastric precursor lesions and cancer. Meanwhile, ornithine decarboxylase (ODC) is a key enzyme that catalyzes the formation of polyamines which are critical for cell growth. So far, the possible regulation of ODC by H. pylori and its virulence factors, and the associated mechanism in gastric epithelial cells remains undefined. In the present study, we found that cellular ODC protein was upregulated by wild-type H. pylori infection and ectopic expression of a cytotoxin-associated gene A (CagA). As a negative control, there was no such effect by cagA-mutant H. pylori infection. Results of signal protein inhibitor treatment demonstrated that the Src, MEK (mitogen-activated protein kinase kinase) and ERK (extracellular signal-regulated kinase) pathway was involved. Moreover, when c-Myc was inhibited, the stimulatory effect of CagA on ODC expression was abolished. Clinically, a positive correlation between c-Myc and ODC expression was observed in patient-derived abnormal gastric tissues. These results implied that the Src/MEK/ERK/c-Myc pathway was required for CagA-mediated ODC induction. Finally, inhibition of ODC expression led to decreased foci formation of gastric epithelial cells before and after H. pylori infection, and ODC protein was over-expressed in precancerous gastric lesions and primary gastric cancer. Collectively, our findings provide new insights into the mechanism behind H. pylori-infection-associated gastric diseases.
Keywords
Introduction
Helicobacter pylori (H. pylori) selectively colonizes in the stomach causing chronic inflammation, 1 which increases the risk of chronic gastritis, gastric ulcer and gastric adenocarcinoma. Some H. pylori strains possess a cytotoxin-associated gene (cag) pathogenicity island (cagPAI) that is present in about half of the Western strains and most of the Eastern strains. 2 CagA, a 120–140 kDa virulence protein encoded by cagPAI, is injected into gastric epithelial cells by the type IV secretory system of H. pylori. 3 Subsequently, most of translocated CagA can be tyrosine-phosphorylated at its Glu–Pro–Ile–Tyr–Ala (EPIYA) motif by Src kinase. 4 The phosphorylated and unphosphorylated CagA interact with multiple host proteins to activate downstream signal pathways, such as the Ras/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway, nuclear factor κB (NF-κB) pathway and β-catenin pathway. 5 Eventually, these events induce changes in cell morphology, cell motility, cell proliferation and intercellular junctions. 5 Accordingly, oncoprotein CagA participates in gastric epithelia injury caused by cagA-positive H. pylori infection. Some studies have shown that the presence of CagA is associated with a 5.8- to 20-fold risk of gastric cancer. 6
The hallmark of cancer is unlimited cell proliferation. This is related to perturbed expression of some key proteins involved in the regulation of the cell cycle, differentiation and apoptosis. Previous studies have shown that ornithine decarboxylase (ODC) is critical for cell transformation. 7,8 It is the rate-limiting enzyme catalyzing the biosynthesis of polyamines critical for normal cell growth and differentiation. Various growth factors, carcinogens and oncogenes regulate ODC expression through the Ras/MEK/ERK and PI3K/Akt pathways. 9 Some studies have shown enhanced ODC enzyme activity in precancerous gastric lesions and gastric cancer specimens in comparison with normal gastric mucosal tissues. 10–13 Furthermore, ODC enzyme activity in H. pylori-positive patients was much higher than that in negative controls. Eradication of H. pylori infection with antibiotics led to a significant decline in ODC enzyme activity in chronic gastritis and gastric ulcer. 14–16
Although studies have shown that ODC mRNA was over-expressed in gastric cancer tissues 17 and eradication of H. pylori in chronic atrophic gastritis decreased the ODC mRNA level, 18 little is known about the mechanism that causes ODC enzyme activity enhancement by H. pylori and which H. pylori component is responsible for enhanced ODC activity. ODC is a transcriptional target of c-Myc; 19,20 hence, it is necessary to make clear whether enhanced ODC activity induced by H. pylori is associated with c-Myc. In the present study, our main goal was to investigate the effect of H. pylori and its virulence factor CagA on ODC expression, and the role of c-Myc in CagA-mediated ODC upregulation in gastric epithelial cells.
Materials and methods
Cell lines, plasmids and cell transfection
The human gastric cancer cell lines AGS and BGC-823 cells, and immortalized epithelial cell line GES-1 were maintained in our laboratory. AGS was cultured in F12 medium (Hyclone, Logan, UT, USA) with 10% fetal bovine serum (FBS). BGC-823 and GES-1 were cultured in RPMI 1640 medium (Gibco, Langley, OK, USA) with 10% newborn bovine serum. All cells were cultured at 37°C with 5% CO2–95% air. Wild-type cagA/pcDNA3.1(+) plasmid (WT-cagA) and its phosphorylation-resistant plasmid derivative (PR-cagA) were kindly provided by Yongliang Zhu (Zhejiang University, Hangzhou, China). These two plasmids were characterized as described previously. 21,22 Transfection of WT-cagA into gastric epithelial cells could express functional carboxyl terminal CagA (C-CagA) that can be phosphorylated, 21 whereas expressed PR-CagA could not be phosphorylated. 22 A c-myc siRNA expression plasmid was constructed using the pSUPER RNAi system (OligoEngine, Seattle, WA, USA ). We used Fugene® HD transfection reagent (Roche, Indianapolis, IN, USA) to transfect the plasmids into cells according to the manufacturer's instruction. The pcDNA3.1(+) (Invitrogen, Carlsbad, CA, USA) and pSUPER plasmids were used as the control.
H. pylori and bacterial infection
The standard H. pylori strains 26695 and NCTC 11637 (cagA-positive) were used. H. pylori 26695 has EPIYA-ABC type CagA, while H. pylori 11637 has EPIYA-ABCCC type CagA. Moreover, the isogenic 26695 cagA mutant strain was constructed using strain 26695 by insertional mutagenesis. All H. pylori strains were grown in Brucella broth containing 5% FBS with 120 rpm under microaerophillic conditions (5% O2, 10% CO2, 85% N2) at 37°C, harvested by centrifugation, quantified by OD600 and added immediately to cell culture at a bacteria-to-cell ratio of 100:1.
Patient samples and immunohistochemistry
We collected tissue specimens from patients with chronic superficial gastritis (CSG, n = 25), chronic atrophic gastritis (CAG, n = 17) and primary gastric cancer (GC, n = 9), who underwent biopsy or gastrectomy. All tissue samples (4 μm) were deparaffinized, rehydrated and antigens retrieved in a microwave oven (0.01 mol/L citrate buffer, pH6.0). After blocking endogenous peroxidise with 3% H2O2, the primary antibody against ODC (Sigma-Aldrich, St Louis, MO, USA) or c-Myc (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was added to the slides and incubated overnight at +4°C. Then biotin-conjugated goat anti-mouse IgG, peroxidise-conjugated streptavidin complex and diaminobenzidine staining were used to visualize antibody binding. Finally, all slides were counterstained with hematoxylin. The pathologist scored the results as the staining intensity×percentage of cells staining at that intensity, categorized as ‘− (score: 0–1)’ no expression, ‘+ (score: 2–3)’ weak expression, ‘++ (score: 4–6)’ moderate expression and ‘+++ (score: 7–9)’ strong expression. The study was approved by the local ethics committee.
Western blot analysis
Total cellular proteins were extracted with lysis buffer, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. The membranes were incubated in the blocking buffer for two hours and probed with specific antibodies against ODC, c-Myc, CagA or β-actin (the latter two from Santa Cruz Biotechnology) overnight at +4°C, followed by horseradish peroxidase-conjugated IgG and developed with enhanced chemiluminescent reagent (Millipore, Billerica, MA, USA) according to the manufacturer's instructions. To determine associated signal pathways involved in CagA-mediated ODC upregulation, one of four signal protein inhibitors (PP1 [20 μmol/L], PD98059 [50 μmol/L], BAY11-7082 [5 μmol/L] [all from Invitrogen] and LY294002 [50 μmol/L] [Upstate, Charlottesville, VA, USA]) was used to preincubate with gastric cells for one hour before transfection of WT-cagA. Cells were harvested and assayed for ODC by Western blot.
Colony formation assay
After co-culture with a green fluorescent protein (GFP)-expressing adenovirus vector harboring ODC antisense RNA (a gift from Dr Wei Wang, Shandong University, Jinan, China) for 48 h at a multiplicity of infection of 50, the BGC-823 cells were co-cultured with H. pylori for two hours. Then, cells were seeded into six-well plates (300 cells/well) and cultured for 10–14 d at 37°C with 5% CO2–95% air. Cell colonies were stained with Giemsa and the colony foci were counted.
Statistical analysis
All experiments were performed in triplicate and representative results are shown. Student's t-test was used to compare data for different treatment groups. The chi-square test was used to analyze differences in clinicopathological variables, and Spearman's rank correlation assay was used to assess the correlation of c-Myc and ODC expression (SPSS version 10.0, SPSS Inc, Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
ODC protein was upregulated by H. pylori infection dependent on CagA
To determine the effect of H. pylori infection on ODC expression, we designed H. pylori infection in human gastric epithelial cells GES-1. Cells were incubated with H. pylori 26695 for one, two, four and eight hours. Western blot results showed apparent ODC protein upregulation at all times points compared with control cells, and the effect was time-dependent (Figure 1a). GES-1 cells incubated with H. pylori 11637 for four hours showed similar ODC protein elevation (Figure 1a). Additionally, similar results were also obtained in AGS cells incubated with H. pylori 11637 and 26695 for four hours (Figure 1a). In contrast, the isogenic 26695 cagA mutant infection could not induce ODC upregulation in AGS and GES-1 (Figure 1a). These results suggested that H. pylori infection induced ODC upregulation in a CagA-dependent manner. To further confirm the role of CagA, we transfected WT-cagA into AGS, BGC-823 and GES-1 cells. Western blot results confirmed that the expressed CagA protein (C-CagA) induced ODC protein upregulation (Figure 1b). Taken together, these results showed that H. pylori infection induced ODC upregulation in gastric epithelial cells, and this effect was dependent on the bacterial CagA protein expression.
Analysis of ornithine decarboxylase (ODC) and c-Myc expression after Helicobacter pylori infection and WT-cagA transfection. (a) ODC protein was upregulated by H. pylori infection. The cells were infected with H. pylori 26695, 11637 or isogenic 26695 cagA mutant, harvested at the indicated times and analyzed for ODC protein by Western blot. (b) Ectopic expression of CagA induced ODC and c-Myc protein elevation. The cells were transfected by WT-cagA, harvested after 24 h, and analyzed for ODC and c-Myc by Western blot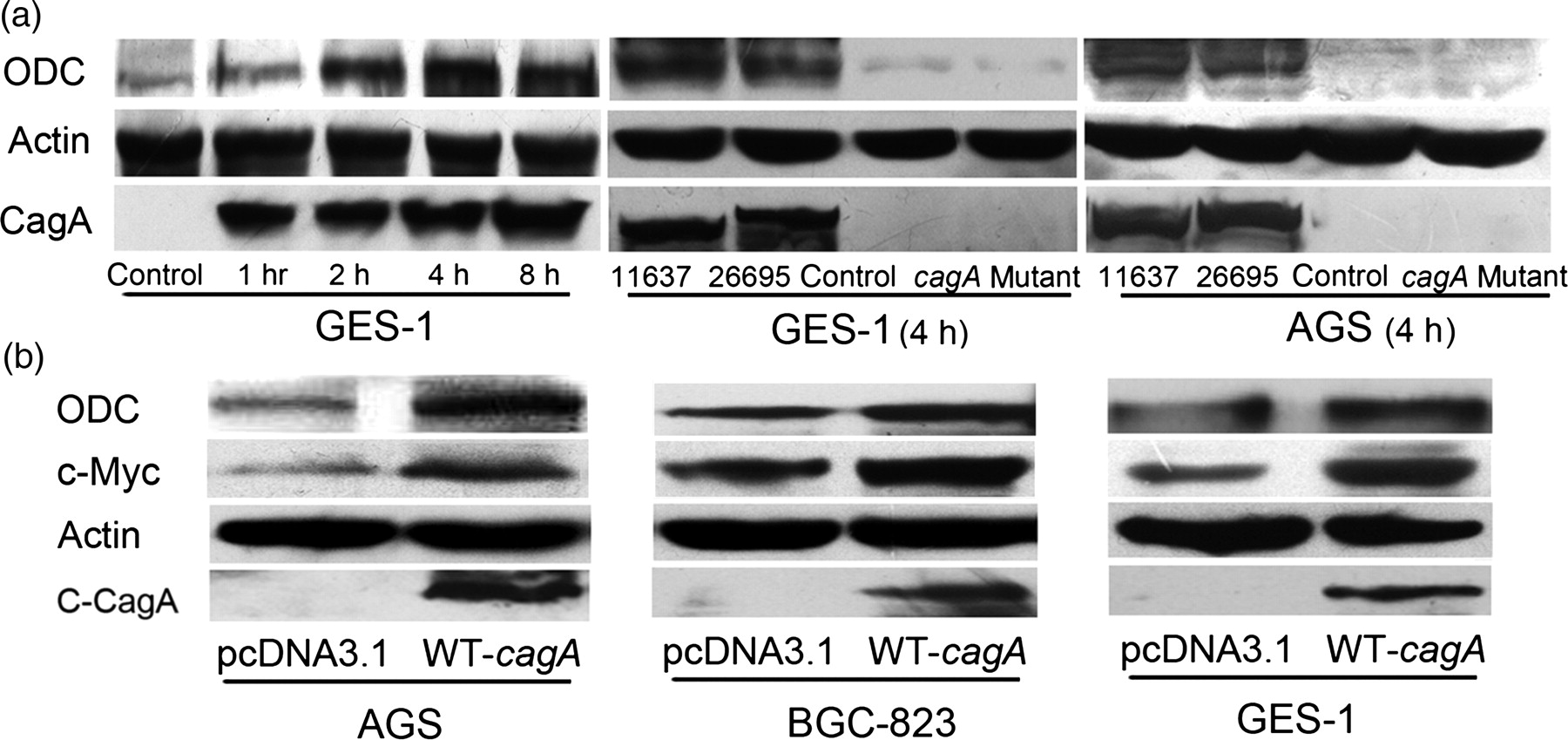
c-Myc was essential for CagA-induced ODC upregulation
To address the potential role of c-Myc in CagA-mediated ODC upregulation in gastric epithelial cells, we performed a number of analyses. First, c-Myc protein was upregulated in the WT-cagA transfected gastric epithelial cells that showed ODC upregulation (Figure 1b). Second, immunohistochemistry (IHC) was used to assay c-Myc and ODC protein expression in abnormal gastric tissues. The pathologist scored the c-Myc and ODC staining in gastric epithelial cells and Spearman's rank correlation assay was used to analyze the IHC scores for c-Myc and ODC in the same gastric tissue. As shown in Figure 2a, c-Myc and ODC expressions were positively correlated (ρ = 0.635, P < 0.001). Accordingly, there was close correlation between c-Myc and ODC in gastric specimens. Finally, c-Myc inhibition by c-myc siRNA expression plasmid resulted in decreased ODC production in BGC-823 (Figure 2b). After c-Myc inhibiton for 48 h, WT-cagA was transfected into the cells. Western blot results showed that CagA-mediated ODC upregulation was attenuated due to c-Myc inhibition (Figure 2c). Together, these results showed that cellular c-Myc protein was essential for CagA-mediated ODC upregulation.
Analysis of c-Myc and ornithine decarboxylase (ODC) in abnormal gastric tissues and WT-cagA-transfected cells. (a) The correlation of c-Myc and ODC expression in abnormal gastric tissues. Spearman's rank correlation was used to assay the score result of ODC and c-Myc immunostaining in the same gastric tissues determined by immunohistochemistry (IHC). (b,c) Attenuation of CagA-mediated ODC upregulation by c-myc siRNA in BGC-823 cells. The cells were transfected by c-myc siRNA before transfection of WT-cagA, harvested after 24 h and assayed for ODC by Western blot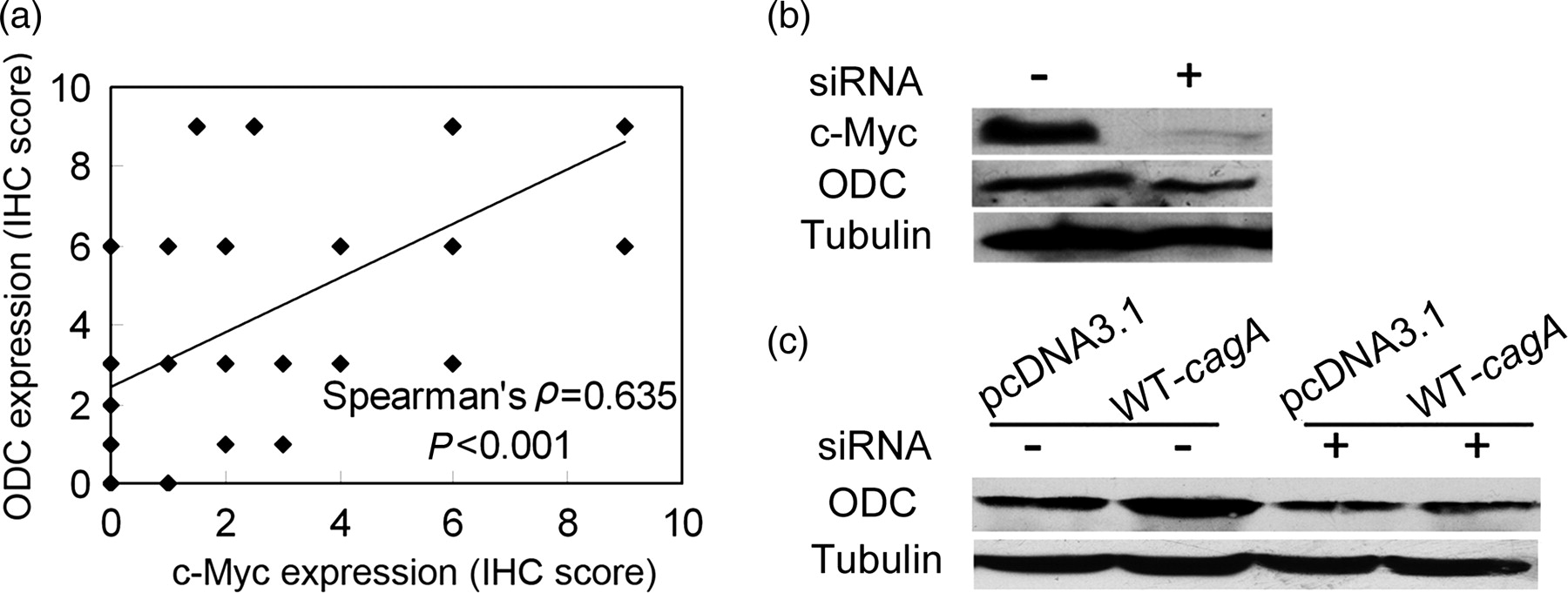
Src, MEK, PI3K and NF-κB were involved in CagA-mediated ODC upregulation
Four signal protein inhibitors (PP1 to block Src kinase, PD98059 to block MEK1 kinase, LY294002 to block PI3K kinase and BAY11-7082 to inhibit NF-κB pathway) were used to elucidate the signal pathways involved in CagA-mediated ODC upregulation. As shown in Figure 3a, ODC upregulation was attenuated after treatment with all the inhibitors. Therefore, the Src, MEK, PI3K and NF-κB pathways were required for CagA-mediated ODC upregulation, and CagA requires phosphorylation. In parallel, transfection of PR-cagA plasmid, a CagA variant that cannot be phosphorylated, could not induce ODC upregulation consistent with PP1 treatment (Figure 3b), confirming the functional requirement of CagA phosphorylation for ODC upregulation.
Effect of signal protein inhibitor and PR-cagA on ornithine decarboxylase (ODC) expression. (a) PP1, PD98059, BAY11-7082 and LY294002 treatment attenuate CagA-mediated ODC upregulation. The BGC-823 cells were preincubated with the indicated signal protein inhibitor for one hour before transfection of WT-cagA, harvested after 24 h and assayed for ODC by Western blot. (b) PR-cagA transfection attenuates CagA-mediated ODC upregulation. The BGC-823 cells were transfected by WT-cagA or PR-cagA, harvested after 24 h and assayed for ODC by Western blot
ODC protein affected the clonogenic potential of gastric epithelial cells and the progression of gastric diseases
To address the effects of ODC protein on cell proliferation, a colony formation assay was performed. GFP-expressing adenovirus vector harboring ODC antisense RNA was used to inhibit ODC protein production and empty adenovirus as the control. Efficient infection of adenovirus and inhibition of ODC production were verified by observing GFP expression under a fluorescence microscope and Western blot (Figure 4a). In comparison with control BGC-823 cells, ODC production-inhibited cells exhibited significantly decreased foci colony before and after H. pylori infection (Figures 4b and c). Therefore, ODC protein inhibition decreased cell proliferation without or with H. pylori infection and ODC protein affected gastric epithelial cell proliferation.
Effect of ornithine decarboxylase (ODC) protein on colony formation potential and gastric diseases. (a) Confirmation of adenovirus infection and ODC inhibition by observing green fluorescent protein (GFP) expression under fluorescence microscopy and Western blot. (b) Diminished colony foci in gastric epithelial cells before and after H. pylori infection. After incubating with GFP-expressing adenovirus harboring ODC antisense RNA for 48 h, the cells were incubated with H. pylori for two hours. Then the cells were seeded into six-well plates, cultured for 10–14 d and stained. (c) The number of colony foci in (a) was counted and analyzed by Student's t-test. (d) Over-expressed ODC protein in gastric lesions determined by immunohistochemistry. Representative results are shown and magnification is ×200. CSG, chronic superficial gastritis; CAG, chronic atrophic gastritis; IM, intestinal metaplasia; AHP, atypical hyperplasia; GC, gastric cancer). (A color version of this figure is available in the online journal)
Immunohistochemical assay of ODC in precancerous gastric lesions and GC
CSG, chronic superficial gastritis; CAG, chronic atrophic gastritis; IM, intestinal metaplasia; AHP, atypical hyperplasia; GC, gastric cancer; ODC, ornithine decarboxylase
*P < 0.05 (assayed by chi-square test), CSG versus CSG with IM, total CSG versus total CAG
Discussion
In the present study we explored the influence of H. pylori on ODC expression using H. pylori to infect gastric cells. A previous study has incubated H. pylori with gastric epithelial cells for 24 h, 25 which implied the feasibility of co-culturing gastric cells and H. pylori up to eight hours. Our studies demonstrated that H. pylori infection induced ODC upregulation dependent on the CagA status of H. pylori but independent of H. pylori strain or cell line (Figure 1a). We used a WT-cagA expression plasmid to confirm the effect of CagA. As expected, the transfected cells showed elevated ODC protein (Figure 1b). All these results confirmed that H. pylori CagA could induce ODC upregulation in gastric epithelial cells. Noticeably, Cheng et al. 26 did not find upregulated ODC in H. pylori-infected gastric epithelial cells, probably due to the different H. pylori strain in their study.
A previous study reported that H. pylori induced ODC expression by c-Myc in macrophages, resulting in macrophage cells apoptosis, 26 but the relationship among H. pylori, especially CagA, c-Myc and ODC in gastric epithelial cells remains undefined. Therefore, we sought to elucidate the role of c-Myc in CagA-mediated ODC upregulation. As shown in Figures 1b and 2a, c-Myc and ODC changed synchronously in the WT-cagA-transfected cells and clinical specimens. This implies the possible regulation of ODC by c-Myc in gastric epithelial cells, which was further confirmed by the results in Figure 2b. When c-Myc protein production was inhibited by c-myc siRNA, CagA-mediated ODC upregulation was attenuated (Figure 2c). Accordingly, we deduced that cellular c-Myc was essential for CagA-mediated ODC upregulation.
To seek for the signal pathways involved in ODC regulation, we used specific signal protein inhibitors. After blocking Src, MEK, PI3K and NF-κB activity, respectively, CagA could not induce ODC upregulation. This implies that CagA exerted its function in a phosphorylation manner (Figure 3a). PR-cagA expression plasmid encoding a variant CagA, which has a tyrosine mutation in the EPIYA motif required for phosphorylation, was used to test our hypothesis. As shown in Figure 3b, PR-cagA transfection could not induce ODC protein elevation in BGC-823. Therefore, CagA required phosphorylation to induce ODC upregulation. Asim et al. 27 also reported that H. pylori-induced c-Myc elevation required phosphorylation and nuclear translocation of ERK. Thus, with the evidence from other studies about the signal pathways involved in ODC regulation, 9,28 we concluded that CagA-mediated ODC upregulation required activating the Src/MEK/ERK/c-Myc and Src/PI3K/Akt pathways. The Src/MEK/ERK/c-Myc pathway mainly regulates ODC transcription because there are two c-Myc binding elements in the ODC gene promoter, and our results confirmed the requirement of c-Myc for CagA-mediated upregulation (Figures 2b and c). The Src/PI3K/Akt pathway mainly regulates ODC translation by regulating phosphorylation of eukaryotic initiation factor 4E (eIF-4E) and its binding protein, 4E-BP1. 9 Further experiments are needed to confirm the function of this pathway in CagA-induced ODC expression. In addition, we found that NF-κB was involved in CagA-mediated ODC upregulation; previous studies have shown that activated NF-κB could induce c-Myc expression, 29 so proceeding studies to define the role of NF-κB in CagA-induced ODC upregulation are needed.
Finally, we found that ODC protein inhibition by ODC antisense RNA diminished colony foci without and with H. pylori infection (Figures 4b and c). Although the percentage of decreased colony foci number in ODC-inhibited cells without infection was higher than those with infection, the results showed that ODC did affect gastric epithelial cell proliferation in an ODC-independent manner. It is known that H. pylori infection could induce numerous proliferation-promoted proteins such as β-catenin. Moreover, ODC protein was over-expressed in precancerous gastric lesions, particularly in chronic gastritis with IM (Figure 4d). Thus, ODC expression was activated in the early stage of gastric diseases, from gastritis to GC, and ODC protein may enhance the progression of gastric lesions. These results imply that H. pylori infection-induced ODC protein elevation could influence the cell proliferation.
In summary, ODC was upregulated by H. pylori CagA through activating the Src/MEK/ERK/c-Myc pathway and may accelerate the progression of gastric diseases, from chronic gastritis to gastric cancer. Conclusively, our findings reveal a novel mechanism underlying H. pylori infection-associated gastric diseases.
Footnotes
ACKNOWLEDGEMENTS
This work was supported by the Natural Science Foundation of Shandong Province, PR China (Nos. ZR2010HQ020, ZR2009CZ001 and ZR2009CM002), National Natural Science Foundation of China (Nos. 81171536, 30972775, 30800406, 30800037, 81001098, 81071313, 81000868, 81172354, 81170514 and 30971151) and National Basic Research Program of China (973 Program 2012CB911202).