
Abstract
PG201, an ethanol extract from a mixture of 12 herbs, has strong antiarthritic activity. To understand the molecular mechanisms underlying its anti-inflammatory effects, PG201-mediated suppression of inflammatory mediators was studied in Raw264.7, a mouse macrophage cell line. PG201 decreased the expression of interleukin (IL)-1β, IL-6 and CC chemokine ligand-2, but not tumor necrosis factor-α, at the protein and mRNA levels in lipopolysaccharide-stimulated Raw264.7 cells. Results from a gel retardation assay indicated that PG201 substantially reduced the DNA-binding activity of the activator protein-1 and cyclic adenosine monophosphate-responsive element-binding protein transcription factors, but not nuclear factor-κB. Western blot and Northern blot analyses showed that PG201 reduced inducible nitric oxide synthase and cytosolic phospholipase A2 (cPLA2) protein expression, but did not affect mRNA expression, ultimately resulting in decreased nitric oxide and prostaglandin E2. The protein expression of cPLA2 was decreased by PG201 in the presence of cycloheximide, an inhibitor of translation, suggesting that PG201 may facilitate the degradation of cPLA2. Taken together, these results suggest that PG201 selectively affects the expression of proteins that play key roles in the inflammatory response at transcriptional and post-translational levels.
Introduction
Osteoarthritis (OA) and rheumatoid arthritis (RA) are chronic inflammatory diseases characterized by the irreversible destruction of the cartilage and subchondral bone by chronic joint inflammation. 1,2 Although factors leading to disease onset are different between OA and RA, inflammatory mediators, such as proinflammatory cytokines, nitric oxide (NO) and prostaglandin E2 (PGE2), play a central role in the pathogenesis of both diseases. 3–9 Tumor necrosis factor (TNF)-α and interleukin (IL)-1β directly induce the synthesis of proteolytic enzymes, such as matrix metalloproteinases (MMPs), which break down macromolecules in the extracellular matrix. 3–5 NO and PGE2 inhibit collagen synthesis and promote the death of articular chondrocytes and synovial fibroblasts in OA and RA. 7,9
A variety of cell types, such as macrophages, fibroblast-like synoviocytes, epithelial cells and mast cells, are involved in the progression of arthritis. 5 Of note, macrophages mainly produce proinflammatory cytokines, chemokines, NO and PGE2 that enter the synovial fluid. 5 It is well known that lipopolysaccharide (LPS), a component of Gram-negative bacterial cell walls, activates macrophages. Once LPS is recognized by Toll-like receptor 4, it induces the phosphorylation of mitogen-activated protein kinases (MAPKs), such as extracellular response kinase (ERK), p38 and jun-N-terminal kinase (JNK), leading to the activation of nuclear factor (NF)-κB, activator protein (AP)-1 and cyclic adenosine monophosphate (cAMP)-responsive element-binding protein (CREB). 10,11 NF-κB, AP-1 and CREB are transcription factors that are critical for the expression of many genes involved in inflammation, such as TNF-α, IL-1β, IL-6, CC chemokine ligand (CCL)-2, inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2. 11–14 Because excessive amounts of inflammatory mediators severely damage tissues, effective blockade of these inflammatory responses is considered to be a critical therapeutic goal. Indeed, many antiarthritic agents, including conventional non-steroidal anti-inflammatory drugs, corticosteroids, disease-modifying antirheumatic drugs and recombinant proteins, such as inhibitors of TNF-α, target these inflammatory mediators. 15,16 However, these agents are not completely effective at treating disease pathology and often cause undesirable side-effects. 16,17 Thus, there is a need for the development of more effective and safer drugs for the treatment of arthritis.
We previously reported that an ethanol extract from a mixture of 12 herbs, PG201, has antiarthritic activities in OA and RA animal models. 18,19 PG201 inhibited the expression of TNF-α and IL-1β in a RA mouse model. In an OA rabbit model, PG201 down-regulated the expression of IL-1β, NO and PGE2, as well as MMPs. PG201 also ameliorated pain in OA patients in a phase II clinical study (manuscript in preparation). However, the effects of PG201 on the expression of inflammatory mediators are not yet clearly defined. In this study, we investigated the molecular mechanisms of PG201 action that govern the regulation of inflammatory mediators using a mouse macrophage cell line, Raw264.7. Treatment with PG201 down-regulated LPS-induced cytokine expression at the mRNA level. A gel retardation assay showed that PG201 repressed the DNA-binding activity of AP-1 and CREB. The expression of iNOS and cytosolic phospholipase A2 (cPLA2), which are involved in the production of NO and PGE2, respectively, was also reduced by PG201 at the protein level. Our data indicate that PG201 may induce anti-inflammatory activities by regulating the expression of cytokine and enzymes at multiple levels.
Materials and methods
Cells and reagents
Raw264.7 macrophages were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal bovine serum (Hyclone, Logan, UT, USA), 100 U/mL penicillin and 100 μg/mL streptomycin. LPS (Escherichia coli, 0111:B4), cycloheximide, dithiothreitol, ethylenediaminetetraacetic acid (EDTA), naphthylenediamine, phosphoric acid, sodium nitrite and sulphanilamide were purchased from Sigma (St Louis, MO, USA). Boric acid, 4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid (HEPES) and Tris base were purchased from Amresco (Solon, OH, USA).
Preparation of PG201
PG201 was prepared using a protocol as described previously. 18 All herbs were purchased from the Kyungdong Herb Market (Seoul, Korea). The moisture content of each herb was less than 10% of its overall weight. The herbs used were as follows: Chaenomelis speciosa Nakai (Rosaceae; Chaenomelis Fructus, 8 g), Achyranthes bibentata Blume (Amaranthaceae; Achyranthis Radix, 8 g), Acanthopanax senticosus Maxim. (Araliaceae; Acanthopanacis Cortex, 8 g), Cinnamomum aromaticum Nees (Lauraceae; Cinnamomi Cortex, 8 g), Gentiana macrophylla Pall. (Gentianaceae; Gentianae Macrophyllae Radix, 5 g), Clematis chinensis Retz. (Ranunculaceae; Clematidis Radix, 5 g), Angelica sinensis Oliv. (Apiaceae; Angelica Radix, 5 g), Cnidium officinale Makino (Apiaceae; Cnidii Rhizoma, 5 g), Gastrodia elata Blume (Orchidaceae; Gastrodiae Rhizoma, 5 g), Carthamus tinctorius L. (Asteraceae; Carthami Flos, 5 g), Phlomis umbrosa Turczaninow (Lamiaceae; Phlomidis Radix, 4 g) and Ledebouriella seseloides Wolff (Apiaceae; Ledebouriellae Radix, 4 g). The herbs (70 g dry weight) were mixed, minced using a grinder (Rong Tong Iron Works, Taichung, Taiwan) and extracted by incubation in 0.7 L of 25% ethanol for three days at room temperature. The supernatant was filtered with a 10 μm cartridge paper, and the ethanol was removed using rotary evaporation (Eyela, Tokyo, Japan). The concentrated extracts were freeze-dried. This process generally produces approximately 10 g of brown powder.
Cell viability
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay kit according to the manufacturer's protocols (Roche, Mannheim, Germany). Briefly, Raw264.7 cells (2 × 104 cells/well) were plated in a 96-well culture plate. Twenty-four hours later, cells were treated with 100 ng/mL LPS and various concentrations of PG201 for 24, 48 or 96 h. Cells were then stained with 10 μL MTT labeling reagent for four hours, followed by the addition of 100 μL solubilization solution and incubation for another 24 h. The absorbance at 570 nm (optical density, OD570–650), at which untreated control cells were taken as 100% of viability, was measured with a microplate reader.
Measurement of cytokines, PGE2 and NO
Raw264.7 cells were plated at 2.5 × 105 cells/mL/well in 24-well culture plates. Twenty-four hours later, the cells were treated with 100 ng/mL or 1 μg/mL LPS and various concentrations of PG201 for 18 or 48 h. The culture supernatant was collected followed by centrifugation at 12,000 ×
NO production was measured in culture supernatant using a modified method as previously described. 20 Briefly, nitrite was measured by adding 100 μL Griess reagent (1% sulphanilamide and 0.1% naphthylenediamine in 5% phosphoric acid) to 50 μL culture supernatant. The OD at 550 nm was measured with a microplate reader. Nitrite concentration was then determined from a sodium nitrite standard curve (0–200 μmol/L). The limit of detection for the nitrite was 3.125 μmol/L.
Western blot analysis
Western blot analysis was performed as previously described. 21 Briefly, Raw264.7 (2.5 × 106 cells/well) cells were plated in 60 mm culture dishes. Twenty-four hours later, the cells were treated with 100 ng/mL LPS and various concentrations of PG201. After 18 h, the cells were washed with cold phosphate-buffered saline (PBS) and lysed using Phosphosafe extraction buffer (Novagen, Madison, WI, USA) for general Western blot analysis. Membranes containing total cell lysate were incubated with antibodies against iNOS (1:4,000; BD Bioscience, Franklin Lakes, NJ, USA), COX-2 (1:250; BD Bioscience), cPLA2 (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and β-actin (1:5,000; Sigma) at 4°C overnight. Membranes were then incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (1:100,000; Sigma) and bands were visualized using ECL (Milipore, Billerica, MA, USA).
Northern blot analysis
Primer sequences used to generate hybridization probes for Northern blot analysis
TNF-α, tumor necrosis factor-α; IL, interleukin; iNOS, inducible nitric oxide synthase; CCL2, CC chemokine ligand 2; cPLA2, cytosolic phospholipase A2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase
Electrophoretic mobility shift assay
Raw264.7 cells (7.5 × 106 cells/well) were plated in 100 mm culture dishes. Twenty-four hours later, the cells were treated with 100 ng/mL LPS and various concentrations of PG201 for six hours. The cells were washed with cold PBS, scraped and harvested. The cell pellet was allowed to swell by adding 200 μL of lysis buffer A (10 mmol/L HEPES [pH 7.9], 10 mmol/L KCl, 1.5 mmol/L MgCl2, 0.1 mmol/L EDTA and 1 mmol/L dithiothreitol) containing complete protease inhibitor (Roche) for 20 min. Two hundred microliters of lysis buffer B (lysis buffer A containing 0.5% Nonidet-P40) was added to disrupt the cell membranes, and the lysate was incubated on ice for 20 min following centrifugation. The pellets containing crude nuclei were washed with 400 μL of lysis buffer A followed by another centrifugation. The pellets were re-suspended in 20 μL of lysis buffer C (10 mmol/L HEPES [pH 7.9], 400 mmol/L NaCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L dithiothreitol and complete protease inhibitor) and incubated on ice for one hour. The nuclear extracts were obtained by means of centrifugation.
The reaction mixture contained 0.01 unit poly(dI–dC), 2 μg salmon sperm DNA, 0.05 pmol [32P]-labeled DNA probe and 10 μg nuclear extracts in binding buffer (Novagen). After incubation for 30 min on ice, the reaction was analyzed on non-denaturing 7% polyacrylamide gel and run with 0.5× TBE buffer (44.5 mmol/L Tris base, 22.3 mmol/L boric acid and 1 mmol/L EDTA [pH 8.0]). Gels were dried, sealed and exposed to autoradiography film at −80°C. Wild-type (WT) and mutant (MT) oligonucleotide sequences designed for probe preparation were as follows (the binding motif is underlined and the lowercase letters indicate mutated nucleotides): AP-1 (WT, GCTTGA
Arachidonic acid release assay
An arachidonic acid release assay was performed using a previously described method.
23
Briefly, Raw264.7 cells (3 × 105 cells/well) were plated in 24-well culture plates. After 24 h, the media was removed and replaced with 1 mL of serum-free DMEM containing 0.1 mCi/mL [3H]-arachidonic acid (NET-298; PerkinElmer). After an overnight incubation, the cells were washed twice with PBS containing 0.1% bovine serum albumin to remove unabsorbed arachidonic acid. The cells were then treated with 100 ng/mL LPS in the presence or absence of 2 mg/mL PG201 for 18 h. After incubation, the culture medium was collected and centrifuged at 10,000 ×
Statistical analysis
Each experiment was repeated at least three times. Results are presented as the mean ± SD of triplicate samples. Comparisons between two groups were analyzed using a Student's t-test. P values less than 0.05 were considered to be statistically significant.
Result
Determination of biological activities of PG201 in cell-based bioassays
PG201 is an ethanol extract prepared from a mixture of 12 herbs. The complexity of PG201, which contains many constituents, makes the preparation of this reagent in a consistent manner very difficult. In this study, the quality of PG201 was standardized by measuring the levels of IL-1β and NO in Raw264.7 cells. The level of these inflammatory molecules was almost undetectable in normal cells, but when treated with LPS, it was dramatically increased. The production of IL-1β and NO was inhibited by PG201 in a dose-dependent manner (Figure 1). The IC50 values of PG201 for IL-1β and NO were 0.73 and 0.87 mg/mL, respectively. Only when the IC50 values from different batches fell within 20% of those from the reference reagent, were the prepared samples deemed permissible for use in the experiments.
Determination of biological activities of PG201 in cell-based bioassays. To measure the effects on IL-1β, Raw264.7 cells were treated with 1 μg/mL LPS and various concentrations of PG201 for 48 h. To measure the effects on NO, Raw264.7 cells were treated with 100 ng/mL LPS and various concentrations of PG201 for 18 h. The levels of IL-1β and NO were measured using an ELISA and Griess assay, respectively. The dotted line indicates the IC50 value for each molecule. Values represent the mean ± SD of triplicate samples from a representative experiment. Three independent experiments were performed. IL, interleukin; LPS, lipopolysaccharide; NO, nitric oxide; ELISA, enzyme-linked immunosorbent assay
Effects of PG201 on the production of proinflammatory cytokines
We have previously found that PG201 could regulate the expression of IL-1β and TNF-α in the mouse RA and rabbit OA models.
18,19
To understand the underlying mechanism, the effect of PG201 on the levels of IL-1β, IL-6, TNF-α and CCL2 were examined in Raw264.7 cells, as these proteins are well-known proinflammatory agents. Cells were treated with LPS and various concentrations of PG201 for 18 h (for IL-6, TNF-α and CCL2) or 48 h (for IL-1β). The levels of cytokines in the culture supernatant were measured by ELISA. The background levels of these cytokines were very low or undetectable, but treatment with LPS dramatically increased their levels. As previously reported, the concentrations used in this study have no effects on cell viability.
18
When cells were treated with PG201, the levels of IL-1β, IL-6 and CCL2 were decreased in a dose-dependent manner. The effect on TNF-α was intriguing as PG201 slightly increased TNF-α production at a low concentration but had little effect at higher concentrations (Figure 2a). The IC50 values of PG201 for IL-1β, IL-6 and CCL2 were 0.73, 1.13 and 0.6 mg/mL, respectively. The protein levels of these cytokines were decreased by up to 90% at 2 mg/mL PG201. These results indicated that PG201 could inhibit the protein expression of IL-1β, IL-6 and CCL2, but not that of TNF-α.
Effects of PG201 on the expression of inflammatory genes in Raw264.7 cells. (a) To measure the effects on IL-6, CCL2 and TNF-α, Raw264.7 cells were treated with 100 ng/mL LPS and various concentrations of PG201 for 18 h. To measure the effects on IL-1β, Raw264.7 cells were treated with 1 μg/mL LPS and various concentrations of PG201 for 48 h. Protein expression of IL-1β, IL-6, CCL2 and TNF-α was measured using an ELISA. Values represent the mean ± SD of triplicate samples from a representative experiment. Three independent experiments were performed. (b) Raw264.7 cells were treated with 2 mg/mL PG201 six hours before (PG201→LPS) or after (LPS→PG201) LPS (100 ng/mL) stimulation, or simultaneously (LPS/PG201). NC indicates untreated cells, while LPS indicates LPS-treated cells. The supernatants were collected at the indicated times after LPS treatment. The level of IL-6 was measured using an ELISA. Values represent the mean ± SD of triplicate samples. (c) Raw264.7 cells were treated with various concentrations of PG201 for 12 h in the presence of 100 ng/mL LPS. Total RNA was prepared followed by Northern blot analysis using [32P]-labeled gene specific probes. GAPDH mRNA was measured as a loading control, and three independent experiments were performed. IL, interleukin; LPS, lipopolysaccharide; NO, nitric oxide; ELISA, enzyme-linked immunosorbent assay; CCL2, CC chemokine ligand-2; TNF-α, tumor necrosis factor-α; GAPDH, glyceraldehyde-3-phosphate dehydrogenase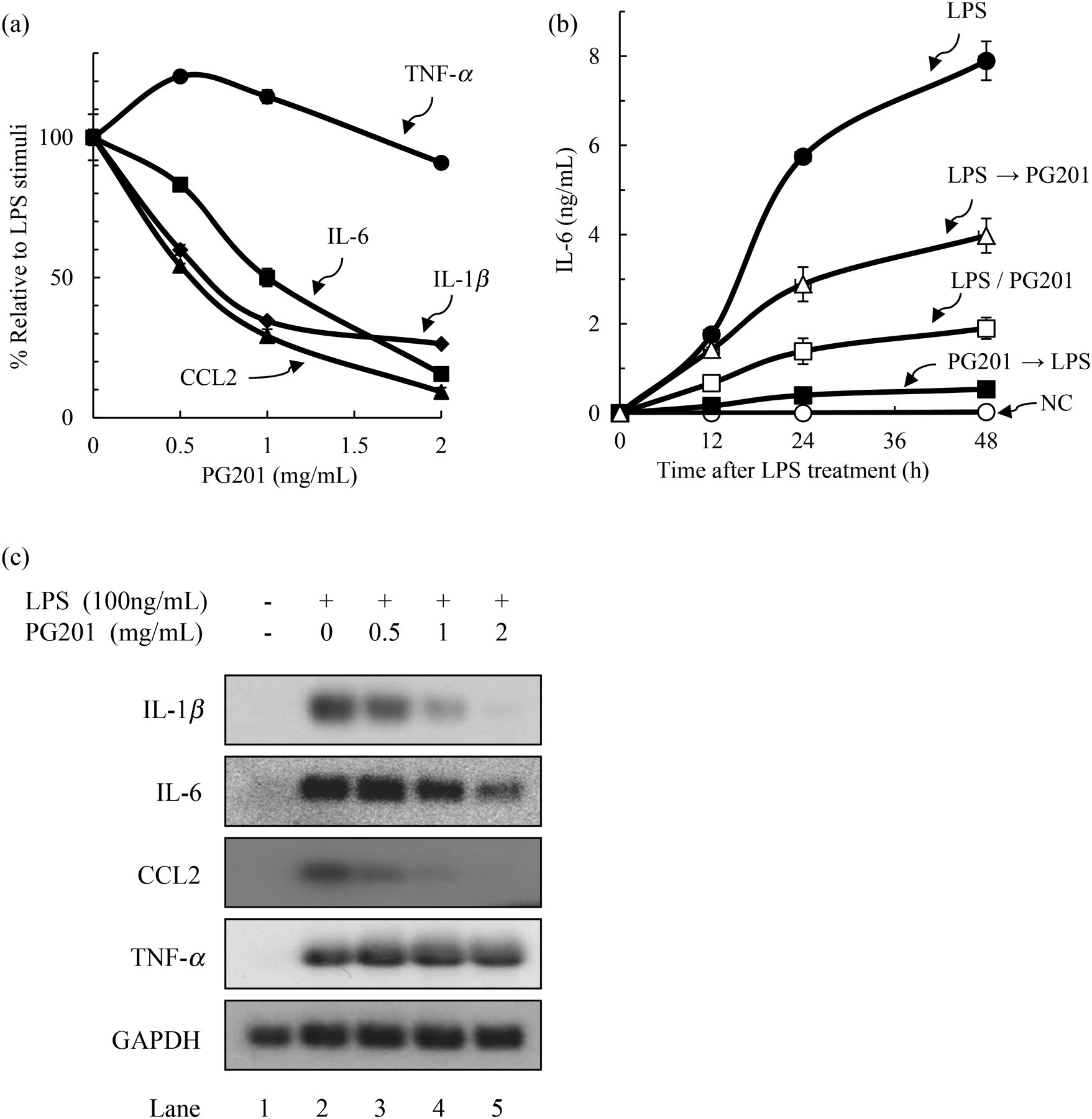
In the above experiments, PG201 was added simultaneously with LPS. To be certain about the effects of PG201, tests were also performed to determine whether PG201 had any effect when treated both before and after LPS stimulation. Raw264.7 cells were treated with 2 mg/mL PG201 six hours before or after LPS stimulation, or simultaneously. The culture supernatant was collected at 0, 12, 24 and 48 h after LPS stimulation, followed by measurement of IL-6. The level of IL-6 was normally undetectable, but dramatically increased to 7.89 ng/mL 48 h following LPS treatment (Figure 2b). Consistent with the above results, the level of IL-6 expression was decreased by 76% when PG201 was added simultaneously with LPS. The pretreatment of cells with PG201 was shown to completely block IL-6 expression. When treated six hours after LPS stimulation, the level of IL-6 was decreased by 50%, so the magnitude of inhibition was lower than both the pre- and simultaneous treatments (Figure 2b). Similar observations were made for other cytokines and also for NO (data not shown). These data suggested that PG201 could effectively inhibit the production of inflammatory mediators in LPS-stimulated Raw264.7 cells.
These cytokines are known to be regulated mainly at the transcriptional level. 11–14 To investigate whether PG201 affects the transcription of these genes, the mRNA level of respective cytokine genes was analyzed by Northern blot hybridization. Raw264.7 cells were treated with LPS and various concentrations of PG201 for 12 h. Total RNA was prepared followed by Northern blot analysis, using a probe specific to each cytokine. The mRNA levels of IL-1β, IL-6, CCL2 and TNF-α were markedly increased by 4- to 12-fold in LPS-treated cells. Consistent with the protein data, PG201 inhibited LPS-induced mRNA production of IL-1β, IL-6 and CCL2 in a dose-dependent manner (Figure 2c). Treatment of cells with 2 mg/mL PG201 decreased the mRNA level of these cytokines by 25-, 2.5- and 18-fold, respectively, while exerting little influence on TNF-α. Taken together, these results show that PG201 could down-regulate the expression of IL-1β, IL-6 and CCL2 at the RNA level.
Effects of PG201 on transcription factors
To understand the molecular mechanisms underlying inhibitory activities on the expression of these cytokines by PG201, it was investigated whether PG201 had any effects on cellular transcription factors involved in inflammation. It is well known that transcription factors, such as NF-κB, AP-1 and CREB, play major roles in the expression of various inflammatory cytokines.
11,14,24–28
The effect of PG201 on these transcription factors was tested by electrophoretic mobility shift assay (EMSA). Raw264.7 cells were stimulated with LPS and various concentrations of PG201 for six hours, nuclear extracts were prepared and EMSA was performed using a specific probe for AP-1, CREB and NF-κB. As shown in Figure 3, the amount of electrophoretically retarded DNA–protein complexes of AP-1, CREB and NF-κB was increased by LPS stimulation (compare lanes 1 and 2 in Figures 2a, b and c). The magnitude of increase was different depending on the transcription factor, with CREB showing the least change. These DNA–protein complexes were specific to each transcription factor because they were effectively competed by the unlabeled wild-type oligonucleotide, but not by respective mutant sequences.
Effects of PG201 on AP-1, CREB and NF-κB transcription factors. Raw264.7 cells were treated with 100 ng/mL LPS and various concentrations of PG201. Nuclear extracts were prepared six hours after treatment. EMSA was performed with [32P]-labeled oligonucleotides targeting AP-1, CREB and NF-κB. The specificity of the DNA-protein complex was verified by competition analysis using unlabeled wild-type (WT) or mutant (MT) oligonucleotides. Three independent experiments were performed. ▸: specific band. EMSA, electrophoretic mobility shift assay; AP-1, activator protein-1; CREB, cAMP-responsive element-binding protein; NF-κB, nuclear factor-κB; LPS, lipopolysaccharide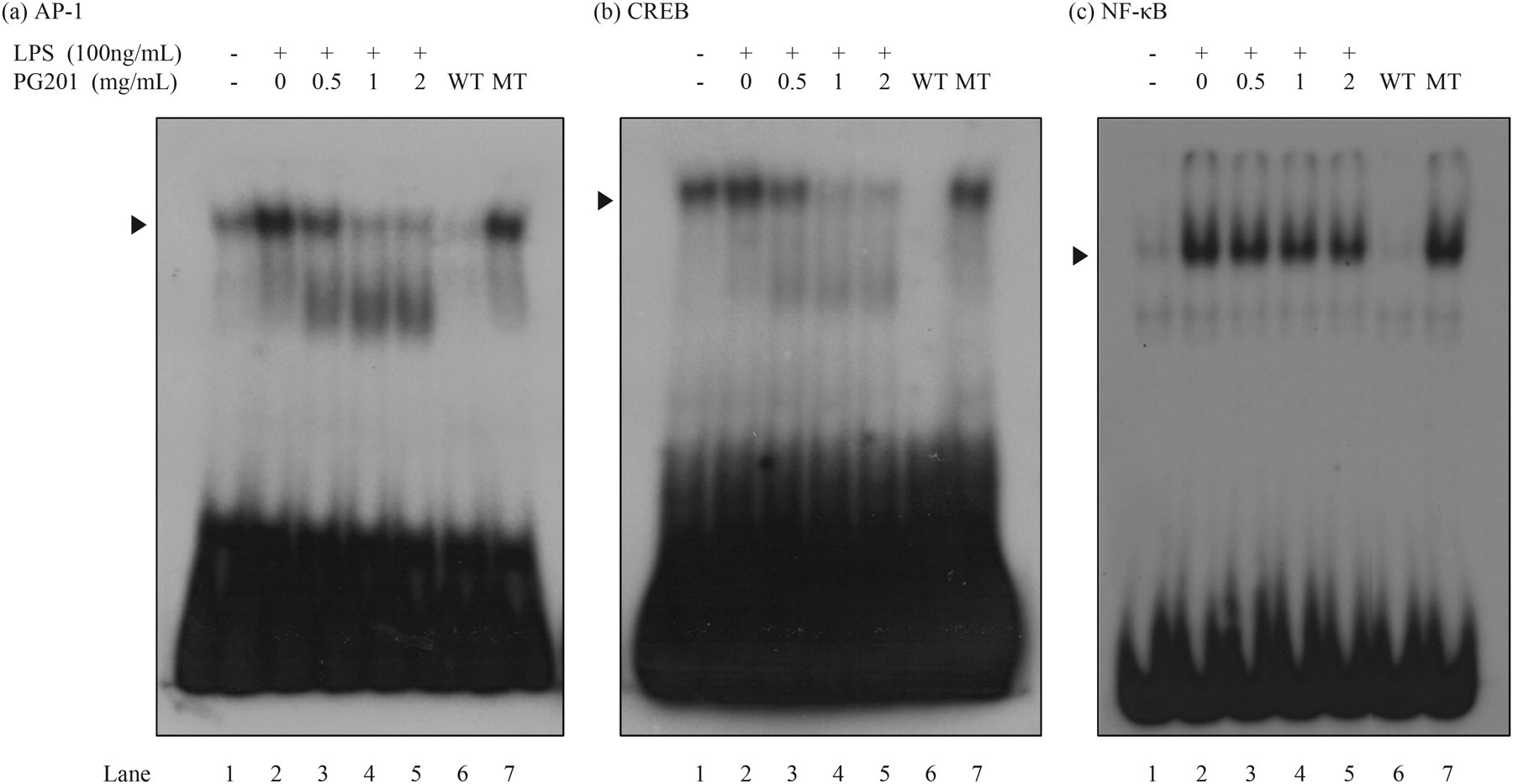
PG201 greatly decreased the DNA-binding activity of AP-1 and CREB in a dose-dependent manner, whereas having little effect on NF-κB (Figures 2a–c). Interestingly, the DNA-binding activity of AP-1 and CREB in PG201-treated cells was reduced to a point lower than the basal level in untreated cells (compare lanes 1, 4, and 5 in Figures 2a, b and c). These data suggested that the inhibitory effects of PG201 on the LPS-induced expression of IL1-β, IL-6 and CCL2 might result primarily from its down-regulation of AP-1 and CREB.
Effects of PG201 on the production of NO and iNOS
NO is an important inflammatory mediator that activates immune cells to produce proinflammatory cytokines and tissue-degrading enzymes, resulting in cell death and cartilage destruction in arthritis.
6,7,29
To test the effect of PG201 on the production of NO, Raw264.7 cells were treated with LPS and various concentrations of PG201 for 18 h, and the level of NO in the culture supernatant was measured by Griess assay. Untreated cells produced a non-detectable level of NO, but treatment with LPS highly increased the production of NO, approximately 18 μmol/L. When cells were treated with PG201, the level of NO was decreased in a dose-dependent manner (Figure 4a). The IC50 value of PG201 for NO was 0.87 mg/mL and the level of NO was reduced by up to four-fold at 2 mg/mL PG201.
Effects of PG201 on the production of NO and iNOS. (a) Raw264.7 cells were treated with 100 ng/mL LPS and various concentrations of PG201. After 18 h, the supernatant was collected and NO was measured using a Griess assay. Values represent the mean ± SD of triplicate samples from a representative experiment. Three independent experiments were performed. (b) Raw264.7 cells were treated with 100 ng/mL LPS and various concentrations of PG201. Total cell lysate was prepared 18 h after treatment. Western blotting was performed using specific antibodies against iNOS and β-actin. β-Actin was measured as a loading control. Three independent experiments were performed. (c) Raw264.7 cells were treated with 100 ng/mL LPS and various concentrations of PG201 for 12 h. Total RNA was prepared followed by Northern blot analysis using a [32P]-labeled probe targeting iNOS or GAPDH. GAPDH mRNA was measured as a loading control, and three independent experiments were performed. LPS, lipopolysaccharide; NO, nitric oxide; iNOS, inducible nitric oxide synthase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase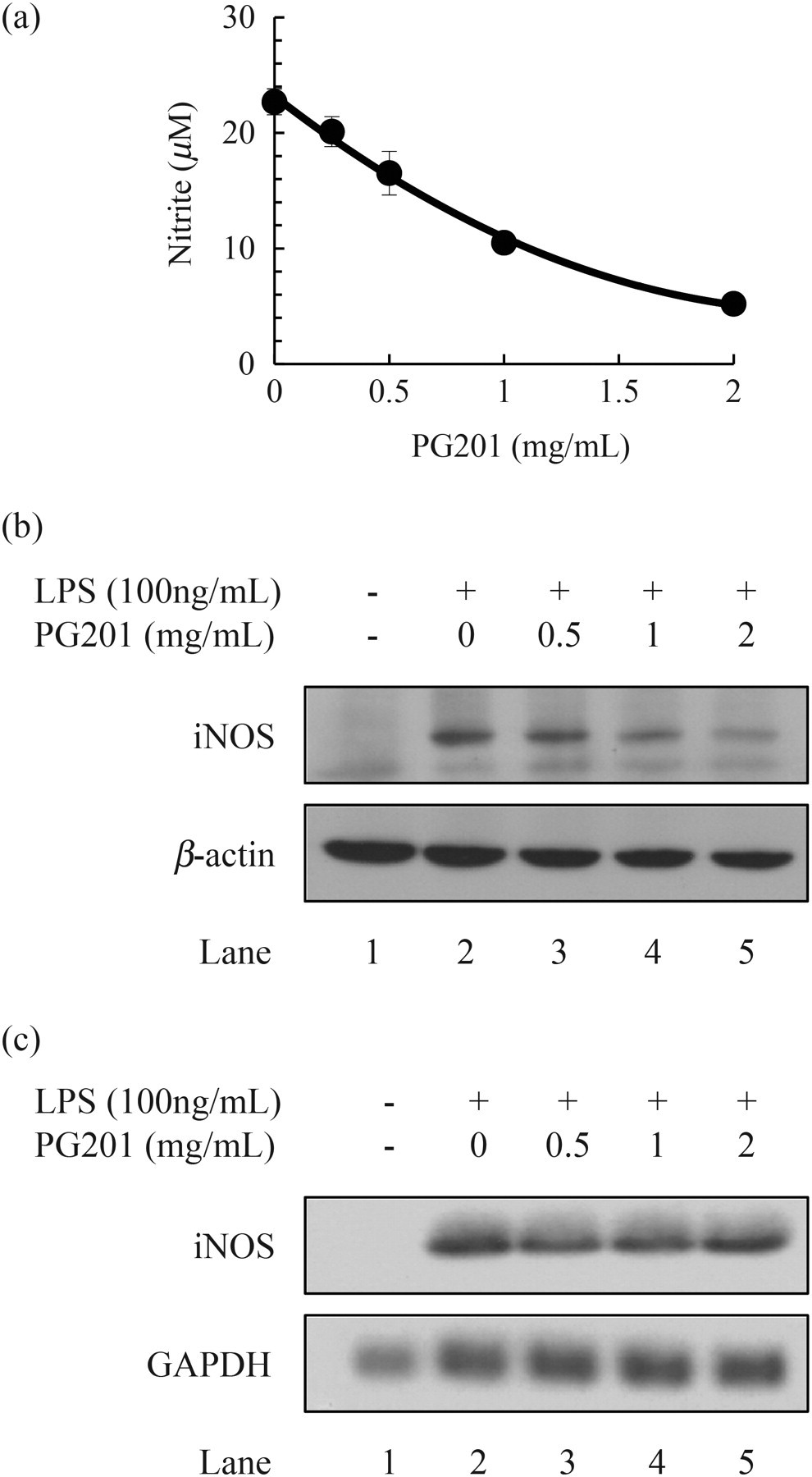
It has been reported that overproduction of NO in macrophage is mainly generated by iNOS. 30 To investigate whether PG201 affects the expression of iNOS, the protein level of iNOS was measured by Western blot analysis. Raw264.7 cells were treated with LPS and various concentrations of PG201 for 18 h. Whole cell lysates were prepared followed by Western blot analysis, using specific antibody to iNOS. In the presence of LPS, the protein level of iNOS was highly increased. However, when cells were treated with PG201, the protein level of iNOS was decreased in a dose-dependent manner (Figure 4b). As it is well known that the expression of iNOS is primarily regulated at the transcription level, 30 the effect of PG201 on iNOS RNA was tested by Northern blot analysis. In contrast to what was expected from the protein data, PG201 did not change the mRNA level of iNOS (Figure 4c). These results indicated that PG201 might inhibit the production of NO by down-regulating the expression of iNOS at the protein, but not at the transcription level.
Effects of PG201 on the production of PGE2
PGE2 is a pathological mediator responsible for the remodeling of cartilage and bone in patients with arthritis. It has been reported that PGE2 inhibits collagen synthesis from chondrocytes and increases the production of MMPs, resulting in cartilage degradation.
8,9
To investigate the effect of PG201 on the production of PGE2, Raw264.7 cells were treated with LPS and various concentrations of PG201 for 18 h, and the level of PGE2 in the culture supernatant was measured by ELISA. When cells were treated with LPS, the level of PGE2 was increased by 17-fold. However, PG201 inhibited this LPS-mediated production of PGE2 in a dose-dependent manner and its IC50 value was 0.54 mg/mL (Figure 5a).
Effects of PG201 on the production of PGE2, cPLA2 and arachidonic acid. (a) Raw264.7 cells were treated with 100 ng/mL LPS and various concentrations of PG201. After 18 hours, culture supernatant was collected and PGE2 expression was measured by an ELISA. Values represent the mean ± SD, and three independent experiments were performed. (b, c) Raw264.7 cells were treated with 100 ng/mL LPS and various concentrations of PG201. Total cell lysate was prepared 18 h after treatment. Western blotting was performed with specific antibodies against COX-2, cPLA2, phospho-cPLA2 and β-actin. β-Actin was measured as a loading control, and three independent experiments were performed. (d) Raw264.7 cells were treated with 100 ng/mL LPS and various concentrations of PG201 for 12 h. Total RNA was prepared followed by Northern blot analysis using a [32P]-labeled probe targeting cPLA2 or GAPDH. GAPDH mRNA was measured as a loading control, and three independent experiments were performed. (e) Raw264.7 cells were pretreated with 100 ng/mL LPS for 18 h. Cells were then washed and treated with 50 ng/mL CHX in the absence or presence of 2 mg/mL PG201. Total cell lysate was prepared at the indicated times. Western blotting was performed with specific antibodies against cPLA2 and β-actin. β-Actin was measured as a loading control, and three independent experiments were performed. (f) Raw264.7 cells were plated in a 24-well plate. The next day, the culture media was removed and replaced with 1 mL of serum-free DMEM containing 0.1 mCi/mL [3H]-arachidonic acid. After an overnight incubation, cells were washed twice with PBS containing 0.1% BSA to remove unabsorbed arachidonic acid. Cells were treated with 100 ng/mL LPS in the presence or absence of 2 mg/mL PG201 for 18 h. After incubation, the culture medium was collected and centrifuged for 10 min at 10,000 × 
The production of PGE2 has been reported to be mainly regulated by phospholipases, cyclooxygenases and PGE2 synthases. COX-2 is well known as a rate-limiting enzyme in PGE2 synthesis. 31 Therefore, the effect of PG201 on COX-2 was first examined. Raw264.7 cells were treated with LPS and various concentrations of PG201 for 18 h. Whole cell lysates were prepared followed by Western blot analysis, using a specific antibody to COX-2. The level of COX-2 was significantly increased by LPS stimulation, but PG201 had no effect on the LPS-mediated increase in the COX-2 protein level (Figure 5b).
It was then investigated whether PG201 could affect the expression of cPLA2 or not. This is because cPLA2 synthesizes arachidonic acid, a substrate of COX-2. cPLA2 (total form) was constitutively expressed in untreated cells, and the level of cPLA2 was not changed by LPS stimulation (Figure 5c). When cells were treated with PG201, however, the level of cPLA2 was dramatically decreased, even at 0.5 mg/mL, the lowest concentration of PG201 (Figure 5c, lanes 2–5). Consistent with the result from cPLA2, phosphorylated cPLA2 was also decreased by PG201 (Figure 5c). These data suggested that PG201 might inhibit the production of PGE2 by down-regulating the expression of cPLA2.
To understand the molecular mechanisms underlying the inhibitory activity of PG201 on the protein level of cPLA2, the mRNA level of cPLA2 was analyzed. Raw264.7 cells were treated with LPS and various concentrations of PG201 for 12 h. Total RNA was prepared followed by Northern blot analysis, using a probe specific to cPLA2. In contrast with the result from protein analysis, PG201 did not change the mRNA level of cPLA2 (Figure 5d).
We next investigated the effect of PG201 on the protein stability of cPLA2. Raw264.7 cells were treated with cycloheximide (CHX), a translation inhibitor, in the absence or presence of PG201 after LPS pretreatment. Whole cell lysates were taken at 0, 3, 6 and 12 h following treatment, and the level of cPLA2 was measured by Western blot analysis. As shown in Figure 5e, the level of cPLA2 was not significantly affected by LPS pretreatment at time 0, and it remained little changed regardless of the presence of CHX (Figure 5e, lanes 1, 2, 3, 5 and 7). When cells were treated with PG201, however, the level of cPLA2 was visibly decreased as compared with that in untreated cells at 3, 6 and 12 h (Figure 5e, compare lanes 3 versus 4, 5 versus 6 and 7 versus 8). These data suggested that PG201 could reduce the protein stability of cPLA2.
To confirm that the PG201-mediated reduction of cPLA2 protein stability affects the production of arachidonic acid and its metabolites, an arachidonic acid release assay was performed. Raw264.7 cells were pretreated with [3H]-arachidonic acid for 24 h and washed with PBS to remove residual [3H]-arachidonic acid. Cells were treated with LPS and PG201 for 18 h, and the radioactivity in the culture supernatant was measured by scintillation counting. Arachidonic acid release was increased in LPS-treated cells by 2.3-fold as compared with that in untreated cells. But when cells were treated with PG201, the level of arachidonic acid release was inhibited by 57% (Figure 5f). Taken together, PG201 appears to decrease the level of PGE2 by reducing the protein stability of cPLA2, and thus lowering the amount of arachidonic acid.
Discussion
Our results indicated that PG201 could regulate the expression of a variety of inflammatory molecules at multiple levels. In Raw264.7 cells, LPS-induced expression of IL-1β, IL-6 and CCL2 was decreased by PG201 at the RNA level, probably at the stage of transcription initiation as PG201 controls the DNA-binding activity of two important transcription factors, AP-1 and CREB. PG201 also lowered the protein level of iNOS and cPLA2, but not their RNA level, resulting in decreases in the production of both NO and PGE2.
IL-1β, produced mainly from macrophages and fibroblasts, stimulates chondrocytes and synovial cells to produce other cytokines (IL-6, IL-8 and leukemia inhibitory factor), MMPs and PGE2. 1,5,32 IL-6 amplifies the IL-1-induced increase of MMPs and inhibits proteoglycan production from chondrocytes. 32 CCL2 recruits T-lymphocytes and monocytes into synovial tissues. 3 Therefore, PG201 has the potential to control both inflammation and cartilage degradation. Indeed, it has previously been shown that oral administration of PG201 decreased the level of IL-1β, while simultaneously preventing the degradation of proteoglycan in the collagenase-induced OA rabbit model. 19
Data from the Northern blot and gel retardation analyses showed that PG201 decreased the expressions of IL-1β, IL-6 and CCL2 at the RNA level, probably by regulating AP-1 and CREB transcription factors. 11,12,14 AP-1 is an important factor for the initiation of chronic inflammatory diseases, such as RA, inflammatory bowel disease and psoriasis, and it controls the expression of many genes involved in RA including MMPs. 24,33 It was reported that AP-1 specific inhibitor could repress the initiation of collagen-induced arthritis. 34 AP-1 and CREB were known to be regulated by MAPKs, such as p38, ERK and JNK. 35,36 However, PG201 did not produce any effect on the LPS-induced phosphorylation of MAPKs (data not shown). The detailed molecular mechanism underlying how PG201 affects AP-1 and CREB remains to be elucidated.
It is intriguing that PG201 inhibits the production of IL-1β and IL-6, but not that of TNF-α, in Raw264.7 cells. The TNF-α promoter contains the binding sites for AP-1 and CREB as well as NF-κB, so theoretically, TNF-α expression is expected to be suppressed by PG201. Furthermore, the protein level of TNF-α was decreased by oral treatment with PG201 in the RA mouse model. It is possible that some factor(s) needed for the PG201-mediated suppression of TNF-α may be absent or present at a limited concentration in Raw264.7 cells. Experiments designed to unravel the dull effect of PG201 on TNF-α are currently under way.
The excessive production of NO and PGE2 contributes to cartilage destruction in RA and OA by inhibiting collagen synthesis and increasing the production of MMPs in chondrocytes. 6–8 PG201 decreased the level of NO and PGE2 in LPS-stimulated Raw264.7 cells by controlling iNOS and cPLA2, respectively, at the protein level. iNOS gene expression is known to be regulated at multiple levels, such as transcription, mRNA stability, translation and protein stability. 37 According to our data from Western and Northern blot analyses, PG201 decreased the expression of iNOS at the protein, but not at the RNA levels (Figures 4b and c). It has been reported that degradation of iNOS protein is facilitated by proteasome or cysteine protease, 38,39 and that transforming growth factor-β increased proteasomal degradation of interferon (IFN)-γ-induced iNOS protein in Raw264.7 cells. 40 Our experiment involving cycloheximide revealed that PG201 had no effect on the stability of the iNOS protein, suggesting that the mechanism underlying PG201-mediated suppression of iNOS is complex (data not shown).
The production of PGE2 is catalyzed by cPLA2, COX-2 and PGE2 synthase in the cascade of reactions. Although COX-2 is well known to be a rate-limiting enzyme in the synthesis of PGE2, PG201 did not affect the expression of COX-2. Interestingly, PG201 decreased the protein level of cPLA2, which produces arachidonic acid, a substrate of COX-2. cPLA2 is constitutively expressed in most cells, especially abundantly in mononuclear phagocytes. 41 A variety of cytokines such as IL-1, TNF, colony-stimulating factor and IFN-γ have been shown to increase the activity and synthesis of cPLA2. Phosphorylation of Ser505 is known to be important in the activation of cPLA2. 41 The involvement of cPLA2 in the production of lipid mediators, such as prostaglandins and leukotrienes, makes it an important pharmacological target for inflammatory diseases, such as RA, lung inflammation, microbial infection, neurological disorder and tumor progression. 41–43 When Raw264.7 cells were treated with LPS, the protein level of cPLA2 was not affected, but its phosphorylation was increased. Consistent with this result, the level of arachidonic acid was increased in LPS-stimulated Raw264.7 cells. PG201 decreased the level of constitutively expressed cPLA2 protein and the production of arachidonic acid. The protein stability analysis showed that PG201 might facilitate the degradation of cPLA2 protein. To our knowledge, this is the first report showing the control of cPLA2 through protein degradation. The molecular mechanism of PG201 on the protein degradation of cPLA2 remains to be elucidated.
Our results suggested that PG201 is a very potent anti-inflammatory agent that effectively suppresses the expression of various inflammatory molecules. Although an active compound(s) of PG201 has not yet been identified, its biological effects have been remarkably reproducible and consistent in various animal models as well as in different in vitro cell culture systems, 18,19 when PG201 is manufactured based on cell-based bioassays involving inflammatory cytokines. Data from the phase II trial indicated that PG201 is not only a very safe agent but also significantly relieves the pain in OA patients (manuscript in preparation). PG201 appears to target multiple targets, containing cartilage protective as well as anti-inflammatory activities, so it may be an ideal therapeutic for complex diseases such as RA and OA. Considering the therapeutic potential of PG201, further extensive investigations are warranted that can identify the active compounds and unravel the underlying mechanism at the molecular level.
Footnotes
ACKNOWLEDGEMENTS
This work has been supported in part by the Oriental Medicine R&D Project Grant 02-PJ9-PG1-CO01-0003 (Ministry of Health & Welfare, Korea) and also by a grant given to SK by ViroMed Co Ltd. We would like to thank Eun-Jung Kwon for her assistance in the preparation of PG201.