
Abstract
Canonical transient receptor potential (TRPC) channel proteins have been identified as downstream molecules in a G protein-coupled receptor signaling pathway and are involved in a variety of cell functions due to their ability to regulate intracellular calcium signaling. TRPC channel physiology has been an increasingly interesting and relevant topic over the last decade, and the outcomes from various studies have advanced our understanding of TRPC function in the normal state. Recently, attention has turned to whether or not TRPC proteins are implicated in diseases. Emerging evidence suggests a significant contribution of several isoforms of TRPC proteins to cardiovascular as well as renal diseases. This review focuses on the implication of TRPC proteins as they pertain to diabetes. We summarize the recent findings by other investigators as well as ourselves and additionally discuss the important role of TRPC proteins in the development of various diabetic complications, such as diabetic nephropathy and diabetic vasculopathy. The underlying mechanisms which contribute to these complications are also outlined. Lastly, we elaborate on the role of TRPC proteins as a potential therapeutic target for treating diabetes-associated diseases.
Introduction
Canonical transient receptor potential (TRPC) channels, a seven-member subfamily of the transient receptor potential (TRP) family of ion channels, are present in a wide variety of tissues. Accumulating evidence demonstrated that all TRPC proteins have both physiological as well as pathophysiological relevance. 1 Physiologically, these non-selective cation channels can function either as receptor-operated or as store-operated (SOC) channels, which upon activation increase the intracellular Ca2+ concentration. As a result, TRPC channels contribute to the function of various systems and organs, including the cardiovascular system, the nervous system, the kidneys and the liver. 1–3 In addition to the physiological roles for these channels, mutation or dysregulation of the channels leads to the development of several different disorders, some of which include focal segmental glomerulosclerosis, diabetic complications, cardiac hypertrophy and impaired cognitive function. 4–8 The widespread effects of TRPC channel dysregulation indicate their critical biological importance.
Effect of diabetes or high glucose (HG) on TRPC expression
TRPC channels and diabetes
Diabetic nephropathy
Diabetic nephropathy (DN) occurs in approximately 40% of all diabetics and is the leading cause of end-stage renal disease in America. 16 Accumulating evidence suggests that DN is one of the most prevalent diseases directly linked to TRPC channel dysfunction. Niehof and Borlak 14 reported a downregulation of TRPC1 mRNA as well as protein in Zucker diabetic fatty rats (an animal model for Type 2 diabetes mellitus), in streptozotocin (STZ)-injected rats (an animal model for Type 1 diabetes mellitus) and in kidney biopsies from patients with DN. In a mouse model (db/db mice) of Type 2 diabetes, Zhang et al. 12 found that TRPC1 mRNA in kidney tissues was slightly reduced after 12 weeks, but significantly reduced by 26 weeks compared with their controls. Additionally, we did not find any significant change in TRPC1 protein expression in freshly isolated glomeruli from rats two weeks after STZ injection. 5 These findings promote the idea that diminished TRPC1 expression occurs in the later stages of DN. Although these studies were descriptive and did not demonstrate a cause-and-effect relationship between the abnormal expression of TRPC1 and kidney dysfunction, the specific location of the TRPC1 gene on human chromosome 3q22–24, a region linked to DN, 17–19 alludes to a possible role in initiating DN. 3,20,21
In addition to TRPC1, TRPC6 may also be dysfunctional in diabetes and the abnormal TRPC6 expression or activity in kidney cells may contribute to the development of DN. Earlier studies from our group have demonstrated that chronic high glucose in glomerular mesangial cells as well as diabetes in STZ rats led to a downregulation of TRPC6 mRNA as well as protein. 5 However, the other TRPC isoforms, such as TRPC1 and TRPC3, did not have significant changes. The decrease in TRPC6 protein expression appeared specific for glomerular tissue because the expression of this protein did not change in the aorta but significantly increased in the whole heart tissue isolated from those STZ rats. 22 Since our study was conducted in rats only two weeks after STZ injection, the decrease in abundance of TRPC6 protein might indicate an early change in the development of DN.
Both TRPC1 and TRPC6 are broadly distributed in the kidneys. 3,21,23 Specifically, both TRPCs play an important role in regulating glomerular filtration by regulating contractile function of the glomerular mesangial cells. 20,22 In diabetes, mesangial contractile function is impaired, and reduced Ca2+ influx is believed to be a major contributing factor to the hypocontractility. 24–26 Our functional studies in cultured mesangial cells have demonstrated that both TRPC1 and TRPC6 participate in agonist-stimulated Ca2+ entry through store-operated and/or receptor-operated mechanisms. 20,22 It is known that different TRPC isoforms can interact with each other to form heteromeric channels. 27,28 We have previously shown that TRPC1 and TRPC6 were co-localized and physically interacted with each other in mesangial cells. 21 Thus, the heterotetramers of TRPC1 and TRPC6 may constitute the native functional channels in glomerular mesangial cells. A reduced number of either TRPC1 or TRPC6 channels or both in mesangial cells would lead to impairment of Ca2+ influx and consequently, disturbs intracellular Ca2+ homeostasis, which eventually results in changes in glomerular hemodynamics in diabetes.
Diabetic vasculopathy
Alteration in the contractile response of blood vessels
Abnormalities in vascular reactivity and function are common in diabetes. 29–31 Dysfunction of both vascular endothelial cells and vascular smooth muscle cells (VSMCs) contributes to the diabetes-associated vascular complications. 30,32–36 Vascular smooth muscle dysfunction is characterized by impaired relaxation to vasodilators or an exacerbated response to vasoconstrictors. 30,32–36 All seven members of TRPC are expressed in endothelial cells and at least four isoforms (TRPC1, 3, 4 and 6) are also found in VSMCs. 37,38 Therefore, abnormal function or expression of a particular TRPC in vascular tissues could lead to blood vessel dysfunction. A recent study by Chung et al. reported that both TRPC1 and TRPC6 protein expression in saphenous veins from patients with Type 2 diabetes were significantly decreased; however, their mRNA levels remained unchanged. TRPC4 mRNA expression was elevated in the diabetic vessel; however, its protein level was not significantly different from that in the non-diabetic saphenous veins. 9 These findings suggest that a post-transcriptional process in TRPCs is altered in diabetic vasculatures. Although TRPC protein expressions were reduced or unchanged, the contraction initiated by cyclopiazonic acid, an activator of store-operated Ca2+ channels, was significantly higher in the saphenous veins from diabetic patients than in the vessels from non-diabetic subjects. The authors deduced that the augmented contraction in the diabetic vessels was attributed to increased channel activity, but not due to an altered number of TRPC channels. This conclusion is highly speculative because whether TRPC proteins are truly and solely the molecular components of store-operated Ca2+ channels remains highly debatable. 6,39,40 Moreover, the authors did not examine STIM1 and Orai1, two proteins that have been acknowledged to play a key role in controlling store-operated Ca2+ entry. 41–43 Changes in protein expression of both STIM1 and Orai1 in diabetes have been reported. 44
Contradictory findings were reported in caudal artery smooth muscle from Goto-Kakizaki (GK) rats, 15 an established model of Type 2 diabetes mellitus by selective breeding from originally non-diabetic Wistar rats. 45 Different from other diabetic animal models, GK rats show a significantly lower systolic blood pressure compared with that in Wistar rats. 46 Consistent with a low systolic blood pressure, the contraction of endothelium-denuded caudal artery strips from GK rats in response to cyclopiazonic acid and cirazoline (α1-adrenoceptor agonist) was significantly depressed when compared with vessels from non-diabetic Wistar rats. 15 Surprisingly, the expression of TRPC1, 4 and 6 was significantly increased in the diabetic caudal artery smooth muscle. Considering that TRPC proteins form homo- and heteromeric channels which have distinct electrophysiological properties, 27,28,47,48 the paradoxical phenomenon (impairment of the store-operated Ca2+ entry-mediated vessel contraction and upregulation of TRPC protein expression) could be interpreted as abnormal assembly of endogenous TRPC proteins in GK rat smooth muscle. However, as the authors stated, the possibility that altered expression of STIM1 and Orai1 also contribute to the impaired vessel contraction in diabetes could not be excluded.
Another contributor to the abnormal contractile response of blood vessels in diabetes is endothelial dysfunction. 49 A Ca2+-dependent nitric oxide mechanism has been proposed to underlie the impaired endothelial function. 50 As candidates of receptor-operated and/or store-operated Ca2+ channels in endothelial cells, 37 TRPCs may be associated with the pathological changes in diabetes by disturbing Ca2+ homeostasis in vascular endothelial cells. A recent study by Bishara and Ding 10 demonstrated that among the four TRPC isoforms expressed in cultured bovine aortic endothelial cells (TRPC1, 3, 4, and 6), only the TRPC1 protein level was significantly increased in response to chronic treatment with high glucose, and this higher level of TRPC1 protein was associated with an augmented Ca2+ influx induced by Ca2+ store-depletion. 10 Although a link between the high glucose-induced increase in TRPC1 protein/Ca2+ entry and endothelial dysfunction cannot be established with certainty, this study provides a clue that TRPC1 is a potential therapeutic target for the treatment of vascular complications of diabetes.
Atherosclerosis
Diabetes is an independent risk factor for the development of atherosclerosis, clinically resulting in coronary heart disease, increased risk of cerebrovascular disease and severe peripheral vascular disease. 51,52 In diabetes, impairment of the endothelium, aggregating platelets, adhesion and migration of monocytes to the vascular wall, and migration and proliferation of smooth muscle cells are all key factors for the initiation and progression of atherosclerosis. 53,54 It has been demonstrated that diabetes alters TRPC protein expression in vascular myocytes. In cultured aortic smooth muscle cells from diabetic GK rats, Evans et al. 13 reported that TRPC4 protein was detected at significantly greater levels, but TRPC6 protein was dramatically reduced compared with that in the cells from non-diabetic Wistar Kyoto (WKY) rats. There was no difference in TRPC1 or TRPC5 expression between the two groups. 13 Functional experiments demonstrated that Ang II-induced Ca2+ influx was significantly enhanced in the GK aortic smooth muscle cells and that this Ca2+ entry response was not affected by inhibition of voltage-operated Ca2+ channels. Instead, this Ca2+ entry was depressed by oleoyl-2-acetyl-sn-glycerol, an activator of TRPC3/6/7 channels and an inhibitor to TRPC4/5 channels. 55 Since TRPC3 gene expression was undetectable in both WKY and GK VSMCs and TRPC6 protein was significantly downregulated in GK, the authors concluded that the elevated Ca2+ influx in the diabetic GK aortic VSMCs was attributed to overactive TRPC1/4/5 channels. Their findings in the VSMCs from diabetic aorta are consistent with earlier reports in VSMCs of atherosclerotic vessels where a shift in Ca2+ channel expression occurred with voltage-operated Ca2+ channels downregulated and TRPC protein expression increased. 56,57 Presumably, this shift in Ca2+ channel expression is associated with dedifferentiation of VSMCs from their normal contractile phenotype to a synthetic, phenotypically modulated cell, which is a key step in atherosclerosis. This is also true for atherosclerotic coronary artery disease, which has high prevalence and severity in patients with diabetes mellitus. 53,58 Using Ossabaw swine as an animal model of metabolic syndrome (pre-diabetes), Edwards et al. showed that metabolic syndrome increased TRPC1 (also STIM1) protein expression in coronary VSMCs, which is associated with elevated store-operated Ca2+ entry and greater atherosclerotic coronary artery disease. 59 Exercise training decreased TRPC1 (also STIM1) protein in the smooth muscle cells with a simultaneous decrease in Ca2+ entry and atherosclerosis in the coronary artery. These data suggest an important role for TRPC1-mediated store-operated Ca2+ entry in coronary artery VSMCs in the development of atherosclerosis in diabetes.
As described previously, 10 high glucose significantly increased the abundance of TRPC1 protein in aortic endothelial cells and, consequently, enhanced agonist-stimulated Ca2+ entry. Therefore, altered TRPC channel-mediated Ca2+ signaling may contribute to atherosclerosis by compromising the integrity of the endothelial barrier which permits more direct interaction of the diabetic milieu with smooth muscle. Another role for endothelial cells in the initiation and progression of atherosclerotic lesions is recruitment of circulating monocytes to the arterial intima by upregulation of vascular cell adhesion molecule 1 (VCAM-1) on the endothelial cells. 60 TRPC3 may be involved in the signaling pathway for VCAM-1 upregulation. In human coronary artery endothelial cells, ATP, a stimulus for atherosclerosis, increased the expression of TRPC3 protein. This was in correlation with augmented TRPC3-dependent constitutive and ATP-stimulated Ca2+ influx as well as upregulation of VCAM-1. 61 TRPC3 knock-down resulted in a dramatic reduction of ATP-induced VCAM-1 and monocyte adhesion to the endothelial cells. These findings suggest a potential pathophysiological role for TRPC3 in coronary artery atherosclerosis.
As mentioned above, recruitment of circulating monocytes to the arterial intima is a crucial event in the initiation, progression and fate of the atherosclerotic lesion. Monocyte adhesion to the vascular wall is determined by the interaction between monocytes and the vascular endothelial cells. 60 Therefore, the activity of the monocyte itself also affects the adhesion process. Wuensch et al. 11 recently reported that high-glucose treatment significantly increased TRPC3 and TRPC6 protein expression in human monocytes. The expression of TRPC1 and TRPC5 in monocytes also increased at the mRNA level. Importantly, TRPC6 mRNA, but not TRPC1, TRPC3 or TRPC5 mRNA was significantly higher in monocytes from patients with Type 2 diabetes as compared with control subjects. 11 Since monocyte activation is a Ca2+-regulated process and a key initial event in the pathogenesis of diabetic atherosclerosis, 62–64 elevated TRPC3 and TRPC6 protein expression in monocytes would increase the risk of developing diabetic cardiovascular diseases by enhancing Ca2+ influx.
Platelet hyperaggregability and hyperactivity due to an enhanced Ca2+ entry plays an important role in the development of cardiovascular complications in diabetes mellitus. 65,66 Recent studies have identified several Ca2+-signaling proteins, including TRPC1, 3 and 6 as contributors to agonist-stimulated Ca2+ entry in platelets. 67–70 In platelets isolated from patients with Type 2 diabetes, Zbidi et al., 44 found that TRPC6 protein expression was decreased; 44 however, TRPC3 (also STIM1 and Orai1) protein was significantly upregulated as compared with the platelets from control subjects. There was no difference in TRPC1 protein expression levels between diabetic and control platelets. This study suggests that an upregulation of TRPC3 as well as STIM1 and Orai1 proteins are responsible for the enhanced Ca2+ entry in diabetic platelets, which was reported in their previous observations. 66,71 Another finding reported by Liu et al. 72 showed that TRPC6 protein expression increased in the platelets from patients with Type 2 diabetes mellitus compared with non-diabetic individuals. Treatment of platelets from healthy humans with high glucose increased TRPC6 protein expression on the platelet surface in a time- and concentration-dependent manner, which was accompanied by an increased 1-oleoyl-2-acetyl-sn-glycerol-induced Ca2+ influx, whereas TRPC1, TRPC3, TRPC4 and TRPC5 were unchanged. 72 The exact mechanism for the discrepancy between the two studies is unclear, but could be the differences in protein expression analysis (Western blot versus immunofluorescence), recruitment of diabetic subjects, and severity and stages of diabetes mellitus.
In summary, various TRPC channels contribute to the development of cardiovascular complications of diabetes mellitus by altering Ca2+ signaling in multiple cell types and cell fragments in vasculature and circulation. A diagram depicting a TRPC mechanism for promoting diabetic vasculopathy is presented in Figure 1.
Possible mechanisms for contributions of transient receptor potential (TRPC) channels in vascular cells, circulating monocytes and platelets to diabetic vasculopathy. During diabetes, an upregulation of TRPCs in vascular smooth muscle cells (VSMCs) increases intracellular Ca2+ levels which then promote contraction and possibly proliferation in these cells. In endothelial cells (ECs), an augmentation of TRPC channel-mediated Ca2+ entry impairs the integrity of the endothelium, which consequently increases the permeability of the vascular endothelium. An elevated intracellular Ca2+ level from TRPC channels in ECs also increases the cell surface expression of vascular cell adhesion molecule-1 (VCAM-1), which promotes monocyte adhesion to the endothelium. An increase in TRPC channel-mediated Ca2+ influx in monocytes (M) stimulates the migration and adherence of monocytes to the vascular endothelium, which is a key step for initiation and the progression of atherosclerosis. Furthermore, enhanced Ca2+ entry through TRPC channels in platelets (P) results in hyperactivity and hyperaggregability of the platelets, which facilitates the development of cardiovascular complications. (A color version of this figure is available in the online journal)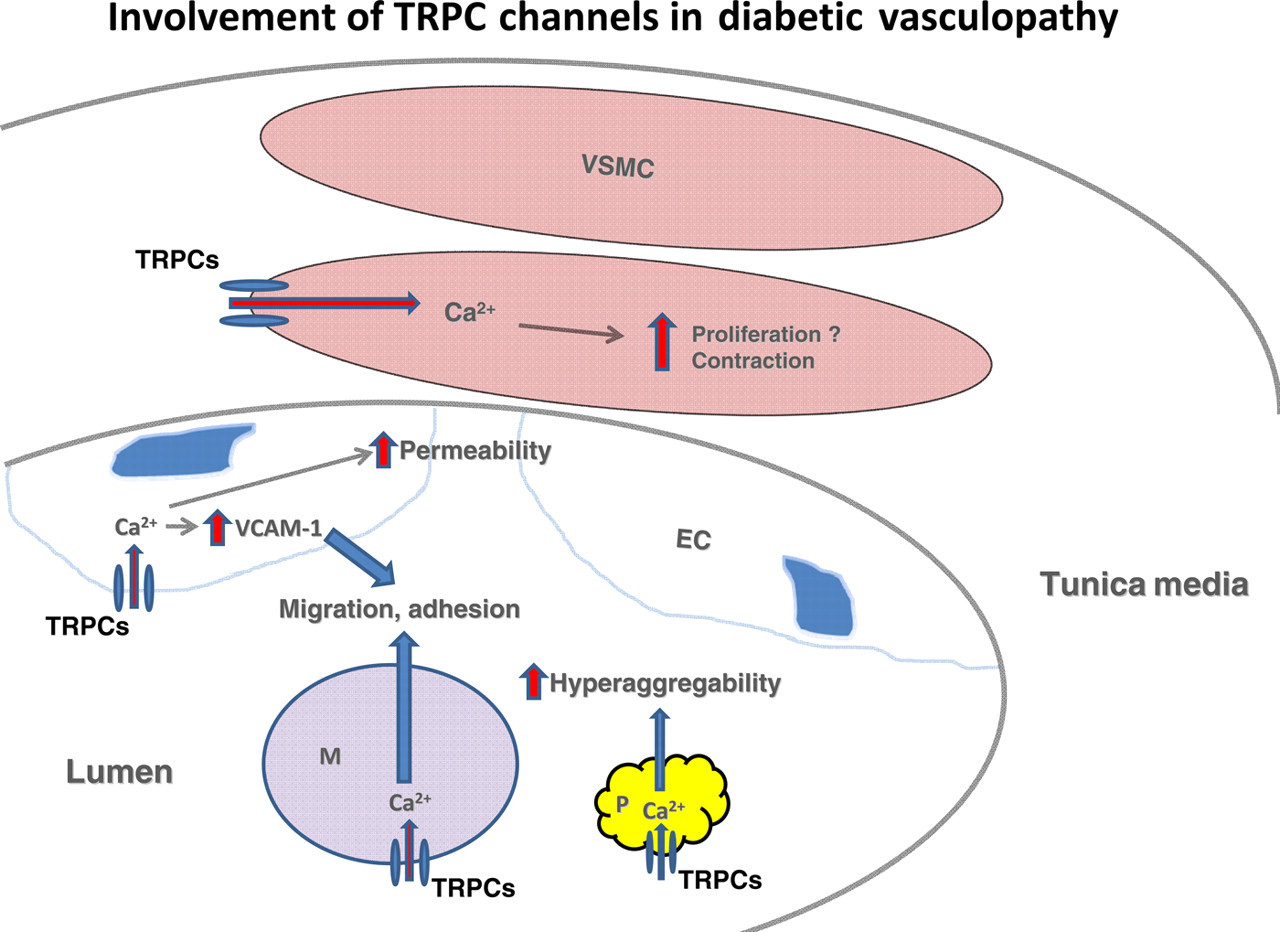
Diabetic neuropathy
Erectile dysfunction in diabetic patients and diabetic animal models is associated with a reproducible autonomic neuropathy and a decline in erectile capacity. 73–75 The diabetes-related erectile dysfunction is presumably related to heightened contractility and/or an impaired relaxation of corporal smooth muscle. TRPC6 channels were recently reported to modulate the tone of the corporal smooth muscle by regulating intracellular Ca2+ concentration. 76 Overexpression of dominant negative TRPC6 channels in human corporal smooth muscle cells significantly reduced the agonist-stimulated Ca2+ response. In vivo gene transfer of dominant negative TRPC6 into smooth muscle of the corpus cavernosum in STZ rats dramatically restored erectile function in those diabetic rats. 76 This study suggests that enhanced TRPC6 channel expression and/or activity in the corpus cavernosum of the penis may underlie erectile dysfunction in diabetes.
Mechanisms involved in TRPC dysregulation in diabetes
Multiple pathways are involved in TRPC channel dysfunction and diabetes since the scope of diabetes and diabetes-related complications is so broad and since the TRPC channels are so diverse in terms of distribution and function. Using pharmacological and molecular biological tools, we have recently proposed a reactive oxygen species (ROS)-protein kinase C (PKC) pathway which contributes to the diabetes-induced TRPC6 protein decrease observed in human mesangial cells. 22 In this cell system, chronic high glucose increases NADPH oxidase activity and/or expression level, which results in overproduction of intracellular ROS. 22 Increased ROS levels then activate PKC, which is capable of either directly or indirectly downregulating TRPC6 in mesangial cells. The PKC family consists of at least 12 isoforms. 77 Mesangial cells are known to express PKCα, βI, βII, γ, δ, θ, ξ and μ. 78–80 It appears that PKCα is one of the isoforms, if not the sole isoform, responsible for mediating the high glucose and ROS effect on TRPC6 expression. An inhibitor (Gö6976) for both PKCα and β, but not an inhibitor (LY333531) for PKCβ alone significantly blocked both the high-glucose- and ROS-induced TRPC6 decrease. 22
Wuensch et al., 11 have also described a ROS mechanism for high-glucose-induced changes in TRPC mRNA or protein expression in human monocytes. Different from our findings in glomerular mesangial cells, elevated ROS levels in monocytes lead to increases in TRPC expression. It is possible that different cell types and conditions could utilize diverging pathways originating at increased ROS levels, which lead to the upregulation or downregulation of a particular TRPC isoform. Over-production of ROS has been previously described as one common pathological process in diabetes as well as in cells cultured in chronic high glucose. 81–84 Additionally, there has been an increasing amount of evidence to support the role of ROS in the development of diabetic complications, such as DN. 85–87 Thus, oxidative stress may be a general mechanism for diabetes-associated TRPC protein dysregulation.
Both Niehof et al . 14 and Zhang et al. have described a pathway whereby a specific gene, hepatic nuclear factor 4-alpha (HNF4α), becomes dysfunctional as a result of exposure to diabetes or high glucose. Due to HNF4α impairment, TRPC1 gene expression is reduced, and the abundance of TRPC1 protein is decreased in kidneys, which ultimately results in the development of DN.
Liu et al. proposed another mechanism whereby diabetes/high glucose alters TRPC expression. They described a pathway where high glucose directly activates a phosphatidylinositol 3-kinase which then promotes TRPC6 channel translocation to the cell surface and thus increases TRPC6 protein expression in the cell membrane. 72 This increased TRPC6 protein expression in the plasma membrane contributes to atherosclerosis and increased thrombotic events, both of which are major vascular complications in diabetes.
Closing remarks
The studies reviewed in this article provide evidence that diabetes is associated with a global change in abundance of TRPC proteins. However, some results from different groups appear to contradict each other. In this regard, it is worth noting that in many instances, the animal species, cell/tissue types, stages and severity of diabetes, and glucose concentrations differ between sets of experiments. Furthermore, there are likely several different pathways mediating the diabetes-related alteration of TRPC expression. These intracellular pathways may be cell-type specific and therefore contribute to the varying TRPC responses to high glucose or diabetes in different types of cells. Also, the mechanism for the diabetes-associated TRPC dysregulation is currently unclear. Although several molecules, such as ROS, have been proposed to be mediators in this pathological process, 14,22,72 how they upregulate or downregulate a particular subtype of TRPCs in diabetes is unknown. Moreover, whether the alteration of TRPC expression is the cause or a result of diabetic diseases is yet to be determined. Nevertheless, as a result of more information becoming known regarding TRPC channels and their role in diabetes, development of specific TRPC inhibitors may be a potential therapeutic option for various diabetic complications. Additionally, treatments utilizing various gene silencing methods for TRPCs or for genes upstream of the proteins could also be useful for diabetic patients. Given the global pandemic of diabetes, searching for additional therapeutic agents is essential to reduce the immense burden of the disease. TRPC channels may be a novel therapeutic target in treating diabetes and its complications.
Footnotes
Acknowledgements
Part of the work described in this article was supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (5 RO1 DK079968-01A2 to the author RM).