
Abstract
Paraquat (PQ) poisoning induces many physiological and histological changes in the human body, but PQ-induced pulmonary fibrosis is most often associated with death. The signaling pathway associated with pulmonary fibrosis is reliant on transforming growth factor-beta 1 (tgf-β 1) activation of Smad3, as evidenced by Smad3-deficient mice being resistant to tgf-β 1-induced pulmonary fibrosis. Thus, we sought to determine whether targeted silencing of Smad3 gene expression could inhibit PQ-induced pulmonary fibrosis in mice. We developed an RNA interference (RNAi) method using short hairpin RNAs (shRNAs) targeting Smad3. The shRNA expression cassettes capable of effectively silencing Smad3 in L929 mouse fibroblasts were transferred to an adenovirus vector and intratracheally administered into mouse lung. Treated mice presented with inhibited Smad3 mRNA and protein and were resistant to PQ-induced pulmonary fibrosis, as evidenced by suppressed expressions of procollagen type I mRNA and hydroxyproline amino acid. Thus, silencing of Smad3 appears to be a promising alternative strategy for the treatment of PQ-induced pulmonary fibrosis.
Introduction
Paraquat (1,1′-dimethyl-4,4′-bipyridinium dichloride; PQ), a defoliant and herbicide substance, has been widely used in agriculture throughout the world since 1962. However, PQ is also a powerful toxin of humans and animals, and its effects on absorption are immediate and so far untreatable. 1 The ease of access, extreme low cost, rapid mode of morbidity and remarkable high mortality (up to 80%) have made PQ an attractive suicide agent in many third-world countries. 2
PQ ingestion is characterized by rapid systemic uptake and leads to multiple organ injury, although PQ effects on the lung are most significantly associated with progression to death. Postmortem examinations of PQ lungs have revealed substantial pulmonary edema, inflammatory cell infiltration, alveoli hemorrhage, fibroblast proliferation and collagen deposition. 3 The clinical manifestation of PQ poisoning has been divided into two phases: acute respiratory distress syndrome and multiple organ failure, the latter usually causing death within a few days. Once pulmonary fibrosis has developed, death usually follows within several weeks. Survivors of PQ poisoning are characterized by restrictive long-term pulmonary dysfunction and relatively poor quality of life. 4
Transforming growth factor-beta 1 (tgf-β 1) is known to induce collagen synthesis and changes of extracellular matrix deposition, both of which play important roles in various lung diseases, such as pulmonary fibrosis. 5 Moreover, tgf-β 1 activity has been described as a ‘switch factor’ that can mediate fibrosis progress, 6 making it an attractive target of molecular manipulation to control the fibrotic pathogenic state. In the early phase of PQ poisoning, high expression levels of tgf-β 1 mRNA and protein have been detected. 7,8 tgf-β 1 has been extensively studied, and its related molecular signaling pathways are relatively well understood. tgf-β 1 coupling to the transmembrane-β-mediated serine/threonine kinase receptors 9 leads to induced phosphorylation of a subset of downstream signaling molecules, including the signal transducing Smad transcription factors. Two receptor-activated Smads, Smad2 and Smad3, bind to the general signal mediator Smad4. This complex then translocates into the nucleus, wherein the Smads regulate transcription of tgf-β 1 responsive genes, ultimately inducing molecular changes that lead to fibrosis. 9 Smad3 protein plays an important role in the development of pulmonary fibrosis. 10 Studies in Smad3-deficient mice revealed that, in the absence of Smad3, pulmonary fibrosis-inducing agents, bleomycin and tgf-β 1, were rendered benign. 11 This finding suggested to us that down-regulating Smad3 expression in vivo might be a useful therapeutic approach for halting pulmonary fibrosis.
RNA interference (RNAi) is a powerful new technique for specifically and robustly inhibiting the expression of a target gene. RNAi blocks gene expression at the stage of translation. 12 Double-stranded RNA is rapidly processed by an RNase III-type enzyme Dicer, creating shorter (21–25 nucleotide) RNA duplexes called small interfering RNA (siRNA). 13 The high efficiency and specificity of siRNA is responsible for the sequence-specific degradation of target RNAs that contain homologous sequences.
In vivo vector-based delivery of short hairpin RNA (shRNA) has been shown not only to avoid detrimental interferon responses of the host system but also permit tissue-specific targeting. 14 This technique, known as gene therapy, has been used successfully in animal model systems and in humans in clinical practice. Adenovirus has emerged as a preferred vector as it can infect most kinds of cells, 15 and has extremely high transfection efficiency for epithelial cells and stromal cells in the lung. 16
Here, we studied the role of Smad3 in PQ-induced pulmonary fibrosis using an shRNA approach in a mouse model system. Our results showed that in vivo silencing of Smad3 effectively attenuated PQ-induced pulmonary fibrosis.
Materials and methods
shRNA screening and vector construction
To generate the Smad3 knock-down vector, one annealed set of oligonucleotides encoding short hairpin transcripts corresponding to Smad3 mRNA (GenBank accession No. NM_016769) was cloned into a pGenesil1.1 plasmid encoding the enhanced green fluorescent protein (EGFP; Wuhan Genesil Biotechnology Co Ltd, Wuhan, China). The efficacy of these constructs in reducing Smad3 transcript levels was tested in cultured cell systems (described below). A corresponding scrambled shRNA plasmid was designed and synthesized for use as a negative control.
Cell culture and transfection
Mice L929 fibroblasts were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China) and used for plasmid transfection. Cells were divided into five groups: the control group (untransduced cells), the negative control group (transfected with scrambled shRNA plasmid) and the experimental (transfected respectively with pGenesil1.1 Smad3-1, pGenesil1.1 Smad3-2 or pGenesil1.1 Smad3-3) groups.
L929 cells were maintained in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum and 1% antibiotics (10,000 IU/mL penicillin and 10,000 mg/mL streptomycin) at 37°C with 5% CO2. For transfection, 2 × 105 cells were seeded into each well of a six-well tissue culture plate. When the cells reached 70–80% confluence (after overnight incubation), the culture medium was aspirated and the cell monolayer was washed once with prewarmed sterile phosphate-buffered saline (PBS). Transfection was carried out using Lipofectamine 2000 reagent (Invitrogen, Shanghai, China), in accordance with the manufacturer's protocol. After 48 and 72 h of transfection, the cells were examined by fluorescence microscopy to detect EGFP expression. The transfection rate was determined by flow cytometry. Total RNA and protein were obtained for use in realtime reverse transcription polymerase chain reaction (RT-PCR) and Western blot analysis to detect the expression of the Smad3 at different time points to confirm the transfection efficiency of Smad3 shRNAs and inhibition of Smad3 gene expression.
Recombinant adenovirus production
The effective shRNA cassettes were excised from the pGenesil-1 plasmid and transferred to a pGSadeno adenoviral expression vector (Wuhan Genesil Biotechnology Co Ltd). The adenovirus was packaged, recombined and amplified. The recombinant adenovirus was validated by observing the fluorescence. Control recombinant adenovirus carrying the scrambled shRNA was constructed in the same manner. The adenovirus-shRNA vectors were purified by cesium chloride ultracentrifugation. Purified viruses were dialyzed in PBS with 10% glycerol and stored at −70°C until use.
Animals and drug administration
All procedures involving animals were approved and performed in accordance with the guidelines from the institutional Ethics Commission. C57BL/6J mice (male 8–10 weeks old, weighing 18–22 g) were obtained from the animal facility of the China Medical University. They were housed under controlled environmental conditions, with 12 h illumination per day and free access to standard rodent feed and water. To induce pulmonary fibrosis, mice (n = 72) were injected intraperitoneally with 10 mg/kg PQ (Sigma, St Louis, MO, USA). Treated mice were then randomly divided into four groups (n = 18 each), for therapeutic intervention: PBS; non-target, to receive 50 μL dosage of 1 × 109 plague-forming unit (pfu) adenovirus containing scrambled shRNA; and adeno-shRNA2 and adeno-shRNA3, each receiving 50 μL dosage of 1 × 109 pfu adenovirus containing two different preparations of shRNA targeting the Smad3 gene. After two hours of PQ treatment, the interventions were injected intratracheally into the lungs of mice. In each group, six mice were sacrificed on days 0, 7 and 28 after treatment for analysis. The time just before the PQ administration was designated as day 0.
Specimen collection and tissue preparation
Mice were sacrificed by exsanguination via the abdominal aorta. Left lobes of the lungs were collected. One-half was fixed in 4% paraformaldehyde, embedded in paraffin wax, sectioned and stained with hematoxylin–eosin. The remaining left lung tissue was frozen at −70°C for future assessment of hydroxyproline content. The right lung tissue (30 mg) was snap-frozen in liquid nitrogen and stored at −70°C for future RNA extraction. On days 0 and 7, an equal amount of right lung tissues were obtained and frozen at −70°C for future detection of nuclear Smad3 protein by Western blot.
Determination of hydroxyproline
The hydroxyproline content in the lungs was assayed by the use of a commercially available hydroxyproline detection kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), following the manufacturer's instructions.
Total RNA preparation and qualitative RT-PCR
Total RNA was extracted from cells or lung tissues by using the RNAiso Reagent (Baoxin Biotechnology Co Ltd, Xi'an China), according to the manufacturer's instructions. cDNA was generated by using the SYBR PrimeScript RT-PCR kit (Baoxin Biotechnology Co Ltd), and qualitative PCR (qPCR) was carried out on a Prism 7500 Fast PCR System (Applied Biosystems, Inc, Foster City, CA, USA). The mRNA expressions were assayed by using the following primers: tgf-β 1, (sense) 5′ TTTCCGCTGCTACTGCAAGTC 3′ and (antisense) 5′ AGGGCTGTCTGGAGTCCTCA 3′; Smad3, (sense) 5′ CTGGCTACCTGAGTGAAGATGGAGA 3′ and (antisense) 5′ AAAGACCTCCCCTCCGATGTAGTAG 3′; procollagen type I, (sense) 5′ CAGGGTATTGCTGGACAACGTG 3′ and (antisense) 5′ GGACCTTGTTTGCCAGGTTCA 3′; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), (sense) 5′ TGTGTCCGTCGTGGATCTGA 3′ and (antisense) 5′ TTGCTGTTGAAGTCGCAGGAG 3′. Amplification of Smad3 mRNA required an initial denaturation step at 95°C for 30 s. Thereafter, and for all other mRNAs amplified, temperature cycling consisted of 40 cycles of denaturation at 95°C for five seconds, annealing at 60°C for 20 s and elongation at 65°C for 15 s. Transcript levels were normalized by comparison with GAPDH and by using the 2-ΔΔCt method.
Western blot analysis
Transfected cells in culture were harvested at various time points, washed once with cold PBS and lysed in buffer containing protease inhibitors. Lung tissue was mechanically homogenized while in an ice bath, then solubilized in a lysis buffer (20 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L ethylene glycol tetraacetic acid, 1% Triton-X100, 2.5 mmol/L sodium pyrophosphate and 1 mmol/L glycerophosphate) and centrifuged to separate out the cytoplasmic fraction. The nuclear fraction was subjected to ultrasonic wave disruption to break the nuclei. The samples were centrifuged and the supernatants containing nuclear proteins were collected for analysis.
The protein concentrations from both whole cultured cells and lung tissue nuclei were measured with the bicinchoninic acid protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China), using bovine serum albumin as the standard. Samples were then diluted to appropriate concentrations and 30 μg of each were separated on 12% sodium dodecyl sulfate polyacrylamide gels, followed by electrophoretic transfer to a nitrocellulose membrane. Membranes were blocked with 5% blocking reagent in Tris-buffered Saline-Tween (10 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl and 0.1% Tween-20), and then probed with rabbit antimouse Smad3 polyclonal antibody (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. After washing, horseradish peroxidase-coupled sheep antirabbit IgG monoclonal antibody (1:500, Wuhan Boster Bio-Engineering Co, Ltd, Wuhan, China) was applied. The proteins were detected by an enhanced chemiluminescence kit (Pierce, Rockford, IL, USA), according to the manufacturer's instructions. GAPDH was used as an internal control.
Statistical analysis
All statistical analyses were carried out with SPSS 16.0 software (SPSS, Inc, Chicago, IL, USA). Results are presented as the arithmetic mean ± the standard error of the mean. Analysis of variance with Student–Neuman–Keuls method was performed to determine the statistical significance of differences between groups. A P value less than 0.05 indicated significance.
Results
RNAi-mediated suppression of Smad3 expression in L929 cells
Sequences and locations of short hairpin RNAs (shRNAs)
*Hairpin loop sequence
†Antisense sequence
GC, guanine and cytosine
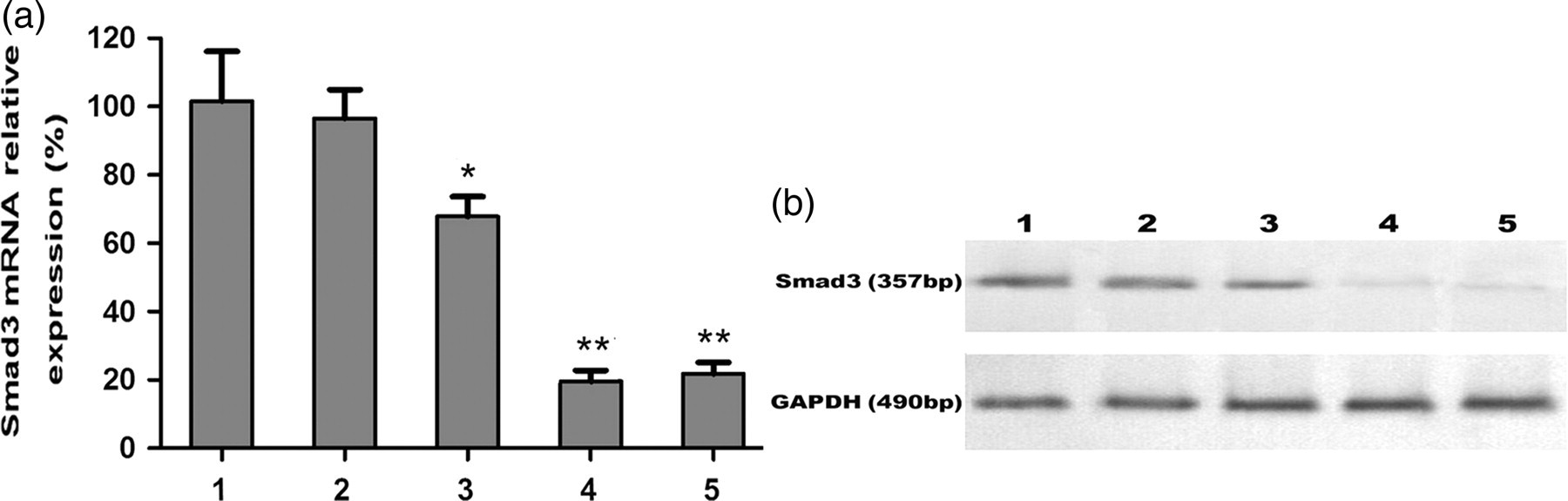
Mice L929 fibroblasts cells were transfected with plasmids containing a human U6 promoter and three Smad3 shRNAs, and the transfection rate was determined by EGFP expression. We showed that a relatively high EGFP expression (85%) was found in cells transfected with Smad3 shRNAs (Figure 1b). Note that little or no EGFP expression (0.62%) was found in cells transfected with control vector (Figure 1a). A significant inhibition of Smad3 mRNA and protein was observed after 48 and 72 h of shRNA transfection. Of the three shRNAs tested, shRNA2 and shRNA3 displayed the highest efficiency for inhibiting expression of Smad3 genes (Figures 2a and b). We therefore used the recombinant adenoviruses containing shRNA2 and shRNA3 for the following in vivo experiments.
Transfection of L929 fibroblasts with short hairpin RNAs (shRNAs) targeting the Smad3 gene. Forty-eight hours after transfection with (a) control or (b) shRNA plasmids, transfection efficiency or positive clones (arrows) were examined under a fluorescence microscope (×200). The transfection rate was determined by flow cytometry as shown in the control (0.62%) and Smad3 shRNA groups (85.12%) The expression of Smad3 mRNA and protein in L929 fibroblasts transfected with pGenesil-1.1 + shRNA vectors. (a) Total RNA was isolated from L929 cells transfected with control or plasmid vectors carrying various Smad3 short hairpin RNA (shRNA) (pGenesil-1.1 + shRNA1, pGenesil-1.1 + shRNA2, pGenesil-1.1 + shRNA3) for up to 72 h. Afterwards, qualitative reverse transcription polymerase chain reaction was performed to detect Smad3 mRNA levels. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. (1) Control empty vector (100%); (2) negative control (97%); (3) pGenesil-1.1 + shRNA1 (67.7%); (4) pGenesil-1.1 + shRNA2 (21.8%); and (5) pGenesil-1.1 + shRNA3 (24.5%). Values are expressed as mean ± the standard error of the mean. *P < 0.05 and **P < 0.01, compared with the controls. (b) Protein was isolated from L9292 cells transfected with (1) control vector; (2) negative control; (3) pGenesil-1.1 + shRNA1; (4) pGenesil-1.1 + shRNA2; and (5) pGenesil-1.1 + shRNA3. Afterwards, Western blot analysis was performed to detect the expression of Smad3 protein. GAPDH was used as an internal control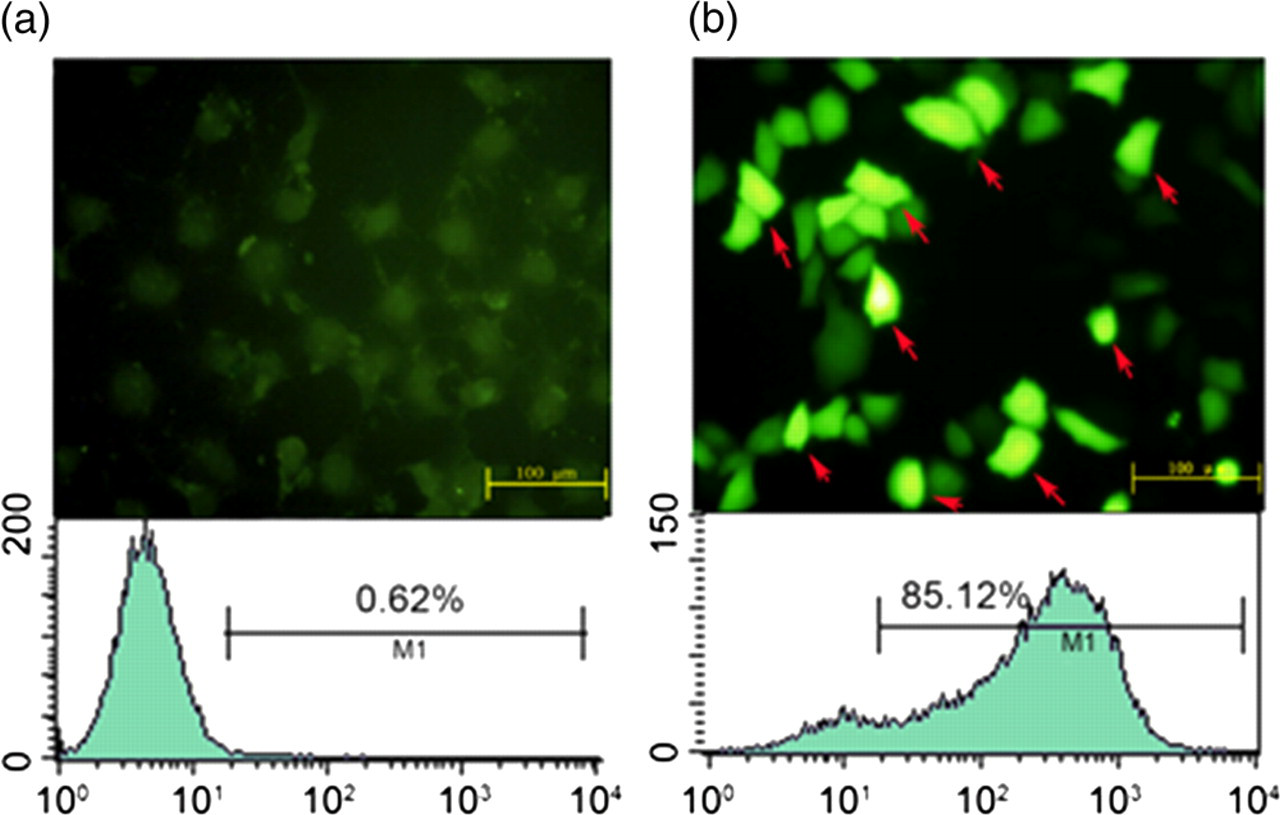
Inhibition of the Smad3 gene expression in vivo by shRNA2 and shRNA3 recombinant adenoviruses
We found that the expression of Smad3 mRNA had decreased in all treated animals on days 7 and 28 as compared with day 0 (data not shown). Compared with the control groups, the expressions of Smad3 mRNA in the adeno-shRNA2 and the adeno-shRNA3 groups were significantly decreased (Figures 3a and b). The difference in Smad3 expression detected between the PBS and the non-target animals had no statistical significance.
The relative expression of Smad3 mRNA on days 7 and 28 in lung tissues. Total RNA was isolated from mice lungs injected intratracheally with control or recombinant adenoviral expression vectors carrying Smad3 short hairpin RNA (shRNA) on days 7 (a) and 28 (b) after paraquat administration. Afterwards, qualitative polymerase chain reaction (qPCR) was performed to detect Smad3 mRNA levels. Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control. Phosphate-buffered saline (PBS), white; non-target, light gray; adeno-shRNA2, dark gray; adeno-shRNA3, black. Values are expressed as mean ± the standard error of the mean. *P < 0.05 and **P < 0.01, compared with the controls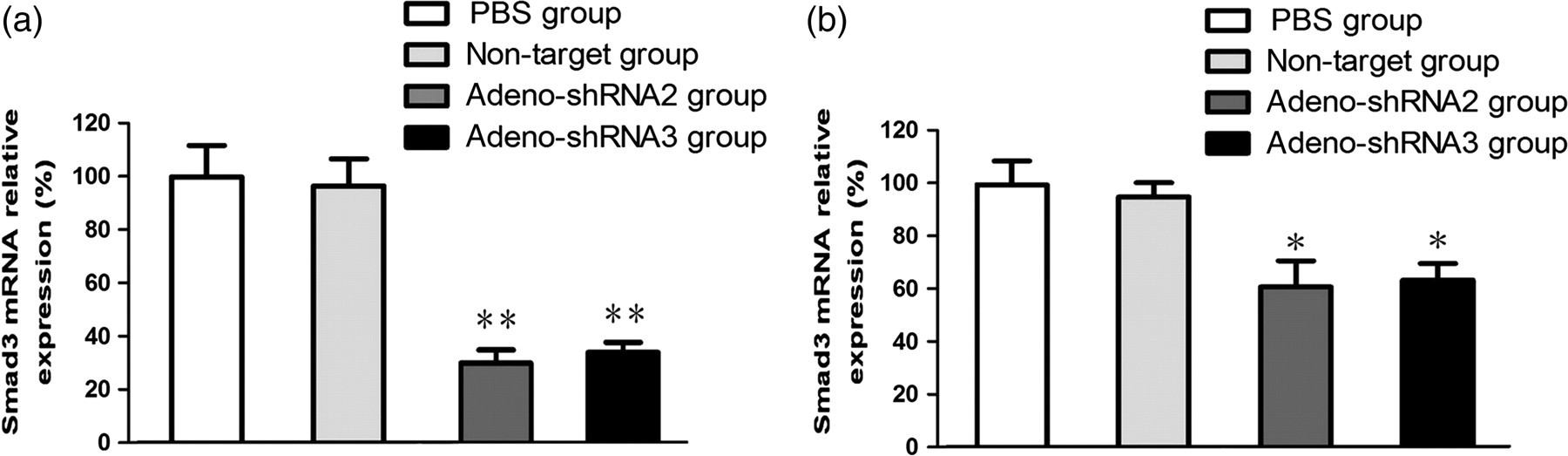
On days 0 and 7, PQ administration caused Smad3 protein to accumulate in the nucleus of PBS and non-target control lung tissues (Figure 4); however, the nuclear expression in the adeno-shRNA2 and the adeno-shRNA3 groups remained relatively unaffected by PQ administration. This finding suggested that functional Smad3 protein was sufficiently inhibited by RNAi-induced silencing of the Smad3 gene.
The effect of short hairpin RNA (shRNA)-silencing on expression of Smad3 protein in the nucleus of lung tissues. Total protein was isolated from the nucleus of mice lungs injected intratracheally with control or recombinant adenoviral expression vectors carrying Smad3 shRNA on days 0 and 7 after paraquat administration. Afterwards, Western blot analysis was performed to detect the expression nuclei Smad3 protein in the lung tissues. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control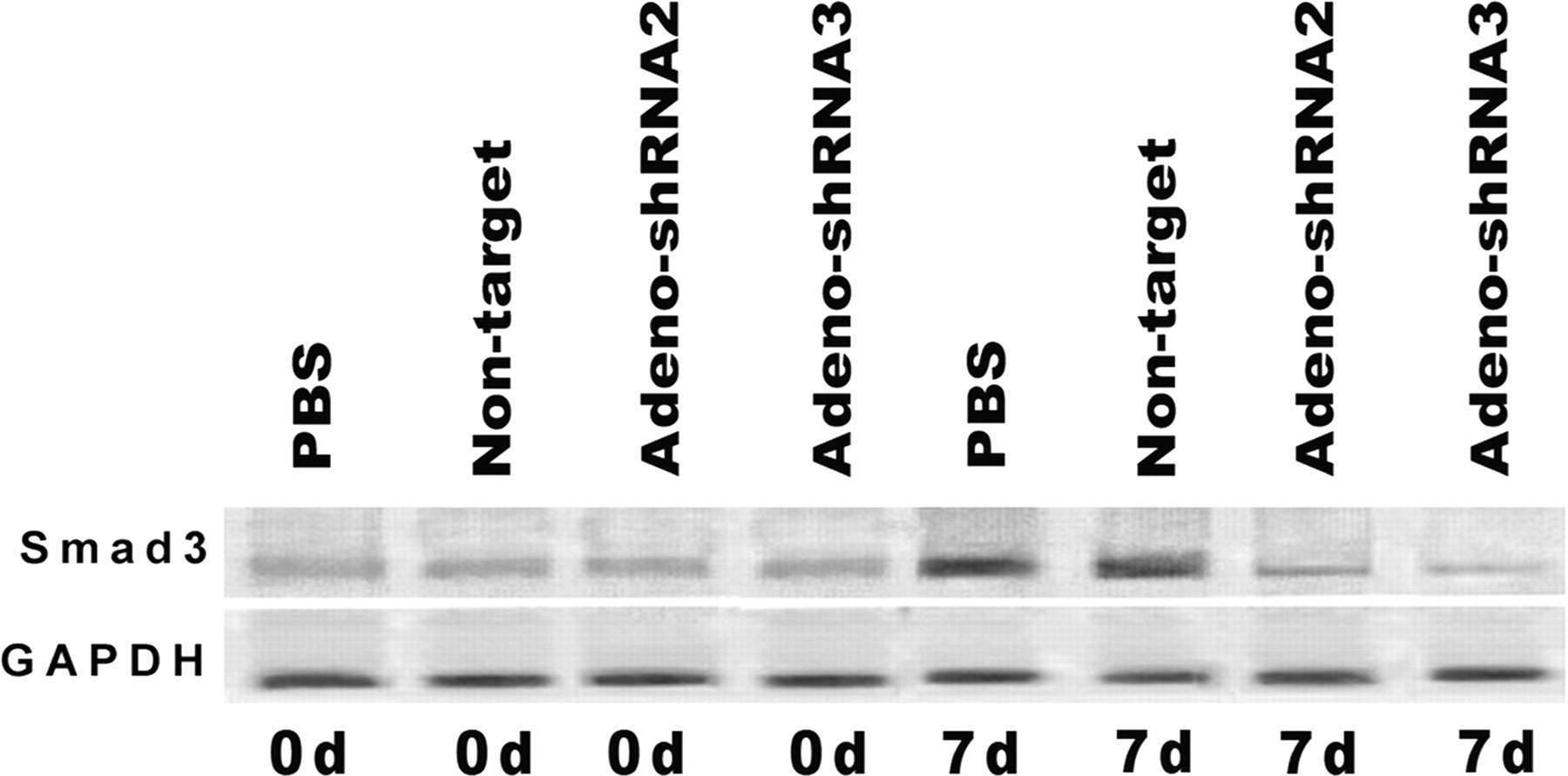
Adenovirus-mediated silencing of Smad3 attenuated PQ-induced pulmonary fibrosis in mice
After seven days of PQ administration, control mice (PBS and non-target groups) presented with obvious histological changes in lung tissues, including expanded and engorged capillaries in the alveolar wall, disorganized alveolus, thickening of the alveolar septum and inflammation (not shown). Examination on day 28 revealed massive proliferation of fibroblasts and wide distribution of collagen fibers (Figures 5a and b). The adeno-shRNA2 and adeno-shRNA3 groups underwent similar histopathological changes in lung tissues during the first seven days of PQ administration (data not shown); however, on day 28, the extent of most changes associated with pulmonary fibrosis was significantly less than those in the control groups, with the notable exception of the thickened alveolar septum. Moreover, the collapse of alveolar space was rare in these Smad3-silenced mice and proliferation of fibroblasts was markedly lower (Figures 5c and d).
The histopathological changes of lungs on day 28 after paraquat administration. Mice lungs were stained with hematoxylin–eosin and microscopically observed at ×400 magnification. The phosphate-buffered saline group (a) and the non-target group (b) exhibited obvious pulmonary fibrosis, such as massive fibroblast proliferation, and wide distribution of collagen fibers. The adeno-short hairpin RNA2 (shRNA2) group (c) and the adeno-shRNA3 group (d) exhibited lower levels of pulmonary fibrosis, fewer instances of collapsed alveolar space (red arrowheads) and less severe fibroblast proliferation (blue arrows)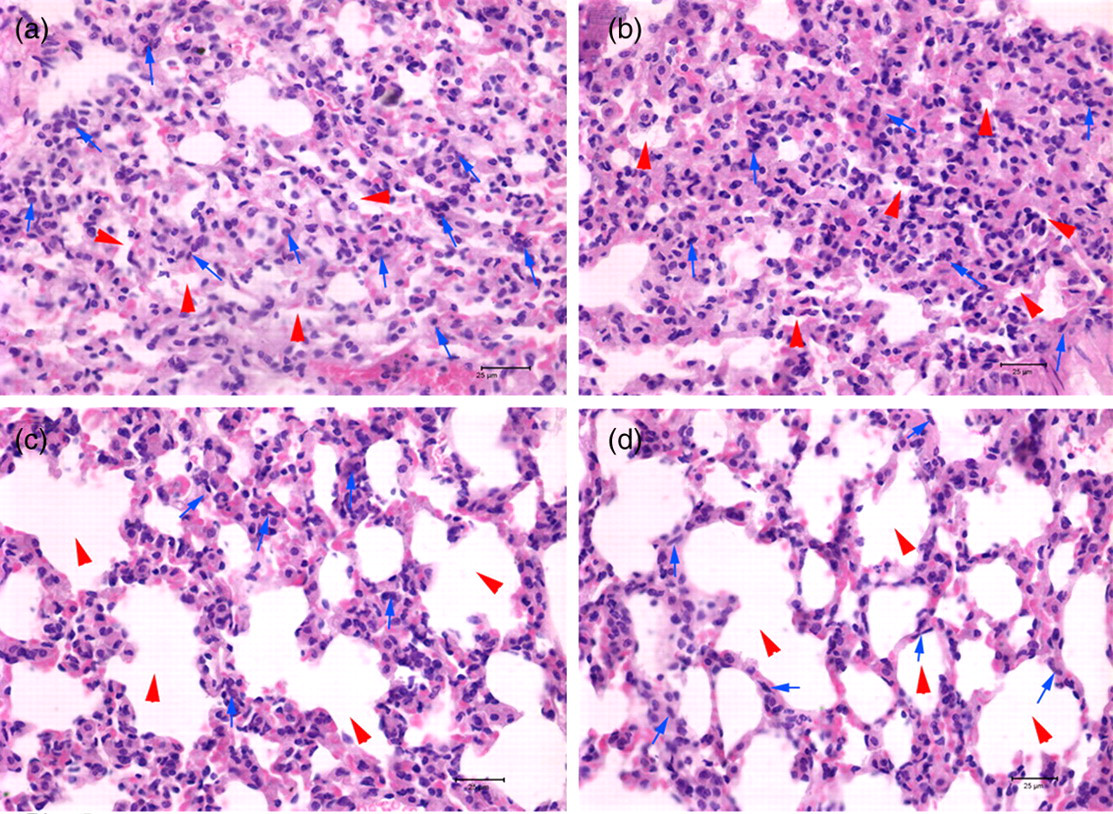
Adenovirus-mediated silencing of Smad3 affected lung cell expression of procollagen type I mRNA but not tgf-β 1
tgf-β
1 mRNA was increased by PQ administration for 7 and 28 days in all groups (Figures 6a and b). There were no statistical differences between any of the tgf-β
1 mRNA levels detected for any of the four groups at the same time points.
The relative expression of transforming growth factor-beta 1 (tgf-β
1) mRNA, procollagen type I mRNA and hydroxyproline content after paraquat (PQ) administration. Total RNA was isolated from mice lungs injected intratracheally with control or recombinant adenoviral expression vectors carrying Smad3 short hairpin RNA (shRNA) after PQ administration. Qualitative polymerase chain reaction was performed to detect tgf-β
1 mRNA on days 7 (a) and 28 (b) and procollagen type I mRNA levels on day 7 (c). Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control. The hydroxyproline content in the lungs on day 28 (d) after PQ exposure was assayed using a commercially available hydroxyproline detection kit. Phosphate-buffered saline (PBS), white; non-target, light gray; adeno-shRNA2, dark gray; adeno-shRNA3, black. Values are expressed as mean ± the standard error of the mean. *Indicates significant difference as compared with the PBS group (P < 0.05)
In contrast, the relative expressions of procollagen type I mRNA were dramatically different among the four PQ-treated groups (Figure 6c). On day 7, procollagen type I mRNA was significantly lower in the adeno-shRNA2 (by 42.6%) and the adeno-shRNA3 (by 58.1%) groups, as compared with that in the PBS group (P < 0.05). No difference was observed between the two control groups (PBS versus non-target group).
Hydroxyproline concentrations were decreased in lungs of Smad3 adenovirus-silenced mice
Changes in whole lung content of collagen, a major extracellular matrix component of fibrosis, were also evaluated by measuring hydroxyproline concentration in the lung tissues of PQ-treated mice. After 28 days of PQ administration, lung hydroxyproline concentrations in the Smad3-silenced mice (adeno-shRNA2: 473 μg/mg and adeno-shRNA3: 526 μg/mg) were significantly lower than that in the non-silenced controls (PBS: 780 μg/mg and non-target: 772 μg/mg) (P < 0.05) (Figure 6d). The difference between the amounts detected in the non-target and the PBS groups were not significant.
Discussion
PQ-induced pulmonary fibrosis responds poorly to known therapeutic interventions, and no currently available drugs can effectively block or reverse progressive fibrosis. PQ intoxication can be lessened by physically removing the poison from the system (via induced vomiting or dilution with intravenous fluids), but extremely rapid uptake and dissemination throughout the body limits the usefulness of such decontamination methods. 17–20 Thus, therapeutic agents inhibiting the ingested PQ are necessary to protect against PQ-induced health effects, such as lung fibrosis.
The tgf-β 1 cytokine is known to play an important role in the occurrence of pulmonary fibrosis by regulating extracellular matrix production and degradation rates, as well as by mediating the expression of a broad range of other signaling molecules promoting the fibrotic response. 5,6,21 tgf-β 1 is also a potent chemoattractant for several immunomodulatory cell types, including monocytes and fibroblasts, and can regulate the function of lymphocytes, monocytes and macrophages. 22–25 tgf-β 1 acts to stimulate the expression of integrins and cell adhesion molecules on the surface of its target cells, 26 thereby enhancing interactions between these cells and the surrounding tissues. Moreover, tgf-β 1 induces monocytes to synthesize and secrete matrix metalloproteinase, such as MMP-2 and MMP-9, which are critical for tissue remodeling. 27
Blockade of the endogenous tgf-β 1 pathway has been proposed as a molecular strategy to ameliorate pulmonary fibrosis. tgf-β 1-neutralizing antibody has been shown to reduce bleomycin-induced pulmonary fibrosis. Using P17, a tgf-β 1 inhibitor peptide, Arribillaga et al. 28 demonstrated that pulmonary fibrosis, including excessive collagen accumulation, was greatly reduced in mice exposed to bleomycin. This result suggested that abrogation of tgf-β 1 signaling was sufficient to offer a therapeutic effect against lung fibrogenesis.
Smad3 expression and activity are key components of fibrosis. During the development of fibrosis, Smad3 protein accumulates in the cell nucleus, where it switches on transcription of the responsive genes of profibrotic cytokine tgf-β 1. Thus, tgf-β 1 and Smad3 determine the mechanism of progressive fibrosis. A previous study indicated that tgf-β 1 activation by adenoviral vector-mediated gene transfer led to progressive pulmonary fibrosis, and this effect was inhibited when the Smad3 gene was knocked out. 29 In addition, another Smad family member, Smad7, has been implicated in the pathogenesis of fibrosis. Intratracheal injection of a recombinant adenovirus expressing Smad7 protected mice against bleomycin-induced fibrosis by a mechanism involving suppression procollagen type I mRNA and reduction of hydroxyproline content. 30 This indicated that gene transfer of a Smad was a feasible approach to prevent cytotoxin-induced pulmonary fibrosis. Recently, it was shown that targeted deletion of Smad3 in mice attenuated the development of fibrotic changes associated with lens epithelial cells after injury. 31 Thus, considering that the tgf-β 1 signaling pathways involving Smads are associated with the pathogenesis of fibrosis, particularly Smad3 with pulmonary fibrosis, we hypothesized that down-regulating Smad3 gene expression in lungs may prevent pulmonary fibrosis.
Our study indicated that the expression of the Smad3 gene was able to be effectively inhibited by RNAi in vivo. This approach was considered biologically safe since other studies have shown that a feedback mechanism regulating Smad3 gene expression exists under cytotoxin-induced conditions of pulmonary fibrosis. That is, reduced Smad3 mRNA corresponded with bleomycin-induced lung injury 32 and baseline levels were recovered upon reinstitution of the normal tgf-β 1 signaling pathway.
The adeno-shRNA2 and adeno-shRNA3 constructed in our study were able to reduce Smad3 expression significantly in mice by day 7, as compared with that in non-silenced control groups. Another previous study found that nuclear accumulation of Smad3 protein in fibrotic lung cells (induced by bleomycin) was highest on day 7. 33 In this study, Smad3 protein accumulated in the nuclear compartments of PQ-induced fibrotic lung cells on day 7, as well, but the accumulation was markedly decreased in tissues with RNAi-induced Smad3 gene silencing. These results collectively indicate the important role of Smad3 in all cytotoxin-induced pulmonary fibrosis.
Collagen accumulation in the lung is the hallmark of pulmonary fibrosis. Many studies have shown that tgf-β 1-activated Smad3 is involved in the expression of the type I collagen gene. 34,35 Wang et al. 36 reported that RNAi-inhibited Smad3 expression led to decreased collagen synthesis in cultured keloid disease fibroblasts. Our study showed that RNAi-inhibited Smad3 gene expression was able to attenuate procollagen type I mRNA expression in an animal model, further confirming the role of Smad3 in mediating collagen expression.
Interestingly, loss of Smad3 did not entirely inhibit pulmonary fibrosis induced by PQ in our animals. While the adenovirus-silenced groups experienced markedly less pulmonary fibrogenesis in response to PQ exposure than their non-silenced counterparts, the Smad3-silenced mice still exhibited some pulmonary lesions, such as thickening of the alveolar septa. One explanation could be that the RNAi method did not completely silence the Smad3 gene. In addition, activities of other signaling pathways might account for the incomplete suppression of tgf-β 1 in the absence of Smad3. Indeed multiple cytokines and growth factors, apart from tgf-β 1, are involved in the development of pulmonary fibrosis, including tumor necrosis factor-α, platelet-derived growth factor, insulin growth factor-1 and interleukins. 37 Thus, it is conceivable that in order to achieve complete inhibition of pulmonary fibrosis, a panel of different cytokines must be manipulated by molecular targeting. Nevertheless, our study showed that pulmonary fibrosis caused by PQ could be attenuated by the RNAi method. RNAi has become a powerful tool for the study of gene function and has been successfully applied as a therapeutic platform. Although issues such as siRNA design, production and delivery still remain to be completely resolved, progress can be expected from further animal studies.
In conclusion, the expression of Smad3 gene is able to be inhibited in vitro and in vivo by RNAi. Moreover, this approach attenuated the formation of PQ-induced pulmonary fibrosis in an animal model. The tgf-β 1/Smad signaling pathway plays an important role in the development of the PQ-induced pulmonary fibrosis and down-regulation of tgf-β 1/Smad3 by RNAi may be a feasible therapeutic approach for pulmonary fibrosis in human PQ-poisoned patients.
Footnotes
ACKNOWLEDGEMENTS
This study was supported by grants from the Specialized Research Fund for the Doctoral Program of Higher Education of China and the Research Foundation of Education Bureau of Liaoning Province, China.