
Abstract
Diadenosine triphosphate (Ap3A) is a vasoactive mediator stored in platelet granules that may be released during coronary ischemia–reperfusion. To study its coronary effects in such circumstances, rat hearts were perfused in a Langendorff preparation and the coronary response to Ap3A (10−7–10−5 mol/L) was recorded. Both at basal coronary resting tone and after precontraction with 11-dideoxy-1a,9a-epoxymethanoprostaglandin F2
α
(U46619), Ap3A produced concentration-dependent vasodilation in the heart, which was attenuated following ischemia–reperfusion. Ap3A-induced relaxation was also attenuated in control conditions and after ischemia–reperfusion by the purinergic P2Y antagonist reactive blue 2 (2 × 10−6 mol/L), the P2Y1 antagonist MRS 2179 (10−5 mol/L), the nitric oxide synthesis inhibitor N-omega-nitro-
Keywords
Introduction
Diadenosine polyphosphates (APnAs) are intracellular and extracellular mediators formed by two adenosine moieties that are linked by two to six phosphate groups. 1 Diadenosine triphosphate (Ap3A), diadenosine tetraphosphate (Ap4A), diadenosine pentaphosphate (Ap5A) and diadenosine hexaphosphate (Ap6A) occur naturally, and they are stored in platelets from which they can be released upon platelet activation. 2 In addition, these APnAs are also found in chromaffin cells 3 and brain tissue. 4 APnAs act as second messengers that mediate insulin release in pancreatic β-cells, 5 and they can also serve as neurotransmitters 6 and extracellular signaling molecules. These mediators have been described in human myocardial tissue 7,8 where they exert both positive and negative inotropic effects on myocardial function. 9 Moreover, at concentrations found in human plasma in physiological conditions (0.1–2 × 10−6 mol/L) 10 , they promote vasodilation in the coronary arteries of rats, 11 guinea-pigs, 12 pigs 13 and dogs. 14
Coronary vascular dysfunction frequently occurs after ischemia–reperfusion, involving reduced vasodilator and increased vasoconstrictor responses. 15,16 These changes may be linked to the clinical phenomenon of no-reflow, 17 a persistent reduction in coronary blood flow after reperfusion. We previously reported that coronary relaxation in response to Ap5A 18 and Ap4A 19 is reduced after ischemia–reperfusion, suggesting that these substances may be involved in the no-reflow phenomenon. Indeed, the concentration of both Ap4A and Ap5A increases in coronary venous blood during ischemia. 20 Ap3A is also present in myocardial tissue 7,8 and increases during ischemia, 20 suggesting that it fulfils a role in the pathophysiology of ischemia–reperfusion. Based on these observations, we analyzed the coronary effects of Ap3A in perfused rat hearts before and after ischemia–reperfusion.
Methods
Experimental set-up
In the present study, 36 male Sprague–Dawley rats were used (300–350 g body weight). All the experiments were conducted in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996) and in compliance with all relevant laws and regulations. The use of these animals was also approved by the Institute's Animal Care and Use Committee. Each heart was used for only one experimental manipulation and five to six hearts were used for each experimental group; therefore, a large number of hearts was used. The hearts were removed from the rats under anaesthesia with intraperitoneal sodium pentobarbital (40 mg/kg) and following intravenous injection of heparin (1000 IU). Next, the ascending aorta was cannulated and the heart was subjected to retrograde perfusion with Krebs–Henseleit buffer (115 mmol/L NaCl, 4.6 mmol/L KCl, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4, 2.5 mmol/L CaCl2, 25 mmol/L NaHCO3 and 11 mmol/L glucose) equilibrated with 95% oxygen and 5% carbon dioxide to a pH of 7.3–7.4. Perfusion was initiated in a non-recirculating Langendorff heart perfusion apparatus at a constant flow rate of 11–15 mL/min to provide a basal perfusion pressure of approximately 70 mmHg. Both the perfusion solution and the heart were maintained at 37°C. Coronary perfusion pressure was measured through a lateral connection in the perfusion cannula and left ventricular pressure was measured using a latex balloon inflated to a diastolic pressure of 5–10 mmHg, both connected to Statham transducers (Statham Instruments, Los Angeles, CA, USA). Left ventricular developed pressure (systolic left ventricular pressure minus diastolic left ventricular pressure), left ventricular end-diastolic pressure, the first derivate of the left ventricular pressure curve (dP/dt) and heart rate were calculated from the left ventricular pressure curve. These parameters were recorded on a Macintosh computer using Chart v 3.6/s software and the MacLab/8e data acquisition system (ADInstruments, Colorado Springs, CO, USA).
Experimental procedure
After a 15-min equilibration period with constant flow perfusion, Ap3A was injected into the perfusion cannula with an infusion pump over three minutes at a constant rate to reach a final concentration of 10−7, 10−6 or 10−5 mol/L. The 10−5 mol/L dose produced a close-to-maximal effect and the response to a 10−4 mol/L dose was only 10–20% higher than that of the 10−5 mol/L dose. Coronary arteries were maintained at a basal resting tone or precontracted with the thromboxane A2 analogue U46619. First, 10−8 mol/L U46619 was added to the perfusion solution and the concentration was increased progressively until a contractile tone of ∼120–140 mmHg was obtained. The concentrations of U46619 required to achieve this effect were 1 × 10−8 to 3 × 10−8 mol/L prior to and 5 × 10−8 to 2 × 10−7 mol/L after ischemia–reperfusion. The response to Ap3A was recorded under control conditions, after which Ap3A and U46619 were washed out and the hearts were exposed to global zero-flow ischemia for 30 min. The hearts were then reperfused for 15 min at the same flow rate used before ischemia, and the response to Ap3A was again recorded at the basal coronary vascular tone or after coronary precontraction with U46619 (Figure 1). The duration of ischemia and reperfusion were chosen on the basis of previous studies demonstrating decreases in the endothelium-dependent coronary relaxation without alteration of endothelium-independent coronary relaxation.
18,21
. Time-control experiments were also performed by administering two successive injections of Ap3A (10−7–10−5 mol/L) separated by 45 min of perfusion without ischemia. As the experiments were performed at a constant flow rate, the coronary perfusion pressure provides a measure of the perfusion resistance and characterizes the contraction or relaxation of the coronary arteries. To assess the stability of the compound, the Ap3A-containing effluent was collected after passing through a heart and infused into another perfused heart in three experiments.
Diagram showing the experimental protocol and the treatment application schedule. 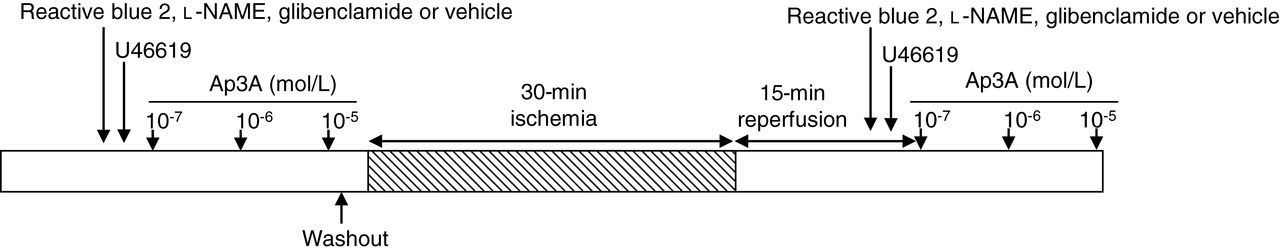
The mechanisms underlying the effects of Ap3A were analyzed by recording the response to this substance after coronary precontraction with U46619 both before and after ischemia–reperfusion, in the presence or absence of: the purinergic P2Y receptor antagonist, reactive blue 2 (2 × 10−6 mol/L); the specific purinergic P2Y1 receptor antagonist, MRS 2179 (10−5 mol/L); the nitric oxide (NO) synthesis inhibitor, N-omega-nitro-
Western blots
To determine the effect of Ap3A on the endothelial nitric oxide synthase (eNOS) and phosphorylated endothelial nitric oxide synthase (P-eNOS) content in the myocardium, four rat hearts were perfused for 15 min with a solution containing Ap3A (10−5 mol/L) injected using an infusion pump. Another four hearts were perfused for the same duration in the absence of Ap3A (control). Immediately after perfusion, the hearts were collected and frozen at −80°C. Total protein from the myocardium was isolated by homogenization of 100 mg of heart tissue in 1000 μL RIPA lysis buffer (50 mmol/L NaH2PO4, 100 mmol/L Na2H2PO4, 0.1% sodium dodecyl sulfate [SDS], 0.5% NaCl, 1% Triton X-100) with 5 mg/mL sodium deoxycholate, 1 mmol/L phenyl-methane-sulfonylfluoride and a cocktail of ethylenediaminetetraacetic acid-free protease inhibitors (Roche Diagnostics, Mannheim, Germany). The lysates were incubated overnight at −80°C and then centrifuged at 14,000 ×
The total protein (100 μg) was resolved on a 10% SDS-polyacrylamide gel under denaturing conditions and electro-transferred to poly(vinylidene difluoride) membranes (Bio-Rad), determining the efficiency of transfer with Ponceau red dye. The membranes were blocked for two hours with Tris-buffered saline (TBS, 20 mmol/L) containing 5% dried non-fat milk and 0.1% Tween 20, and then incubated overnight with agitation at 4°C with the appropriate primary antibodies raised against P-eNOS Ser 1177 (1:200; BD Transduction Laboratories, Franklin Lakes, NJ, USA) and eNOS (1:1000; Abcam, Cambridge, UK). The membranes were subsequently washed and incubated with the corresponding peroxidase-conjugated secondary antibody (Pierce Biotechnology, Rockford, IL, USA), the activity of which was visualized by chemiluminescence (PerkinElmer Life Science, Boston, MA, USA), and quantified by densitometry using a Kodak Gel Logic 1500 Image Analysis system and Molecular Imaging Software, Version 4.0 (Rochester, NY, USA). All blots were reprobed for β-tubulin (Sigma-Aldrich, Madrid, Spain) to normalize each sample for gel-loading variability, and all data were normalized to the control values for each gel.
Statistical analysis
The coronary vascular response is expressed as the mean (±SEM) change in perfusion pressure. Coronary responses before and after ischemia–reperfusion were compared using a paired Student's t-test. The responses in the presence and absence of antagonists were compared using one-way analysis of variance followed by Dunnet's test. A P value of <0.05 was considered significant.
Drugs and chemicals used
The following substances were all obtained from Sigma (Madrid, Spain): P1,P3-di(adenosine-5′) triphosphate ammonium salt (Ap3A), 1-amino-4[[4-[[4-chloro-6-[[3-sulfophenyl]amino]-1,3,5-triazin-2-yl]amino]3–3sulfophenyl]amino]-9,10-dihydro-9,10-dioxo-2-anthracenesulfonic acid (reactive blue 2), 2′-deoxy-N6-methyuladenosine 3′,5′-diphosphate diammonium salt (MRS 2179), 9,11-dideoxy-1a,9a-epoxymethanoprostaglandin F2
α
(U46619), N-omega-nitro-
Results
Effects of ischemia in perfused hearts
Coronary perfusion pressure (mmHg), left ventricular developed pressure (mmHg) and maximal dP/dt (mmHg/s) in perfused rat hearts: before and after 45 min of perfusion without ischemia (time-control); before and after 30 min of ischemia plus 15 min of reperfusion in hearts at basal coronary tone; or after coronary precontraction with U46619 in the presence or absence of reactive blue (2 × 10−6 mol/L), MRS2179 (10−4 mol/L),
n = number of experiments
Significant difference as compared with pre-ischemia–reperfusion values (**P < 0.01)
Significant difference with time-control (# P < 0.05; ## P < 0.01)
Significant difference with respect to hearts at basal tone (†† P < 0.01)
Ap3A response in hearts perfused at basal coronary resting tone
At basal coronary tone, injection of Ap3A (10−7−10−5 mol/L, n = 5) reduced perfusion pressure and this decrease was stronger before ischemia–reperfusion than after (Figure 2). Ap3A induced small fluctuations in left ventricular developed pressure, left ventricular end-diastolic pressure, maximal dP/dt and heart rate which were not statistically significant, both before and after ischemia–reperfusion (Table 2).
Coronary dilation induced by diadenosine triphosphate (Ap3A; 10−7–10−5 mol/L) in perfused rat hearts. Measurements were taken before (control) and after 30 min total ischemia followed by 15 min of reperfusion (left panel), or repeatedly before (first test) and after (second test) 45 minutes of perfusion in time-control experiments (right panel). Left panel: control (•); ischemia–reperfusion (○). Right panel: first test (•); second test (○). Values represent the mean (±SEM) of five experiments. **Significant difference with respect to controls (P < 0.01). Ap3A, diadenosine triphosphate Percentage increase (positive values) or decrease (negative values) in left ventricular developed pressure and maximal dP/dt produced by injection of diadenosine triphosphate (Ap3A, 10−7–10−5 mol/L) before (control) and after ischemia–reperfusion in untreated hearts (control) at basal tone and in hearts precontracted with U46619 in the presence or absence of reactive blue 2 (2 × 10−6 mol/L), MRS2179 (10−5 mol/L),
n = number of experiments Values represent the mean ± SEM
In time-control experiments with hearts perfused at coronary basal tone (n = 4), a second injection of Ap3A 45 min after the first injection had similar effects on perfusion pressure (Figure 2), left ventricular developed pressure, left ventricular end-diastolic pressure, maximal dP/dt and heart rate. Moreover, the Ap3A-containing effluent collected after passing once through a heart produced similar effects to freshly prepared Ap3A when injected into another heart, suggesting that the compound was relatively stable under our experimental conditions.
Ap3A response after coronary precontraction with U46619
When compared with hearts at basal tone, precontraction of the coronary vasculature with U46619 enhanced the Ap3A-mediated decrease in perfusion pressure both before and after ischemia–reperfusion (n = 5). In precontracted hearts, the decrease in perfusion pressure induced by Ap3A was significantly attenuated after ischemia–reperfusion. The effects of Ap3A on left ventricular developed pressure, left ventricular end-diastolic pressure, maximal dP/dt and heart rate were similar in precontracted hearts and those at basal tone, both before and after ischemia–reperfusion (Table 2). We also investigated whether increases in myocardial function and metabolism after U46619 treatment might increase Ap3A-induced vasodilation in this condition. However, increases in left ventricular developed pressure and maximal dP/dt in response to the beta-adrenergic agonist isoproterenol (10−7 mol/L) attenuated the vasodilatory response to Ap3A (data not shown).
Ap3A response in hearts treated with reactive blue 2
Treatment of precontracted hearts with reactive blue 2 (2 × 10−6 mol/L; n = 5) before ischemia–reperfusion attenuated the vasodilatory response to the highest concentration of Ap3A (10−5 mol/L), when compared with that observed in precontracted hearts not exposed to reactive blue 2. By contrast, reactive blue 2 treatment after ischemia–reperfusion attenuated the vasodilatory response to all concentrations of Ap3A used. Following reactive blue 2 pretreatment, Ap3A reduced left ventricular developed pressure and maximal dP/dt without significantly modifying the left ventricular end-diastolic pressure or heart rate, both before and after ischemia–reperfusion (Table 2). Coronary perfusion pressure, left ventricular developed pressure, left ventricular end-diastolic pressure, maximal dP/dt and heart rate were not affected by exposure to reactive blue 2, both before and after ischemia–reperfusion (Table 1).
Ap3A response in hearts treated with MRS2179
In precontracted hearts, MRS2179 treatment (10−5 mol/L, n = 5) before ischemia–reperfusion attenuated the vasodilatory response induced by the highest concentration of Ap3A (10−5 mol/L), when compared with precontracted hearts not exposed to MRS2179. After ischemia–reperfusion, the same dose of MRS2179 attenuated the vasodilatory response to the two highest concentrations of Ap3A used (10−6 and 10−5 mol/L). Moreover, after MRS2179 treatment, Ap3A reduced the left ventricular developed pressure and maximal dP/dt before but not after ischemia–reperfusion, without significantly modifying left ventricular end-diastolic pressure or heart rate (Table 2). The coronary perfusion pressure, left ventricular developed pressure, left ventricular end-diastolic pressure, maximal dP/dt and heart rate were similar in hearts irrespective of whether they were treated with MRS2179, both before and after ischemia–reperfusion (Table 1).
Ap3A response in hearts treated with l -NAME
Before ischemia–reperfusion, Coronary vasodilation induced by diadenosine triphosphate (Ap3A; 10−7–10−5 mol/L) in perfused rat hearts during coronary precontraction with U46619, before (a) and after (b) ischemia–reperfusion, in the presence or absence of reactive blue 2 (2 × 10−6 mol/L), MRS2179 (10−5 mol/L), 
Ap3A response in hearts treated with glibenclamide
In hearts precontracted with U46619, glibenclamide treatment (10−5 mol/L) before ischemia–reperfusion attenuated the vasodilatory effect of the lowest concentration of Ap3A used (10−7 mol/L, n = 6; Figure 3). In precontracted hearts after ischemia–reperfusion, glibenclamide attenuated the vasodilatory response to all concentrations of Ap3A used. In hearts treated with U46619 and glibenclamide, Ap3A significantly increased left ventricular developed pressure and maximal dP/dt without modifying left ventricular end-diastolic pressure or the heart rate, both before and after ischemia–reperfusion (Table 2). Coronary perfusion pressure, left ventricular end-diastolic pressure, left ventricular developed pressure, maximal dP/dt and heart rate were similar in hearts treated with and without glibenclamide, both before and after ischemia–reperfusion (Table 1).
P-eNOS and eNOS content in the myocardium
No differences in eNOS protein levels were observed between control and Ap3A-treated myocardia. However, P-eNOS protein content was significantly higher in Ap3A-treated than in untreated myocardia (P < 0.05) (Figure 4).
Endothelial nitric oxide synthase (eNOS) and phosphorylated endothelial nitric oxide synthase (P-eNOS) protein content in the myocardia of perfused rat hearts treated with (n = 4) or without (control, n = 4) Ap3A (10−5 mol/L) for 15 min. *Significant difference as compared with control group (P < 0.05). Ap3A, diadenosine triphosphate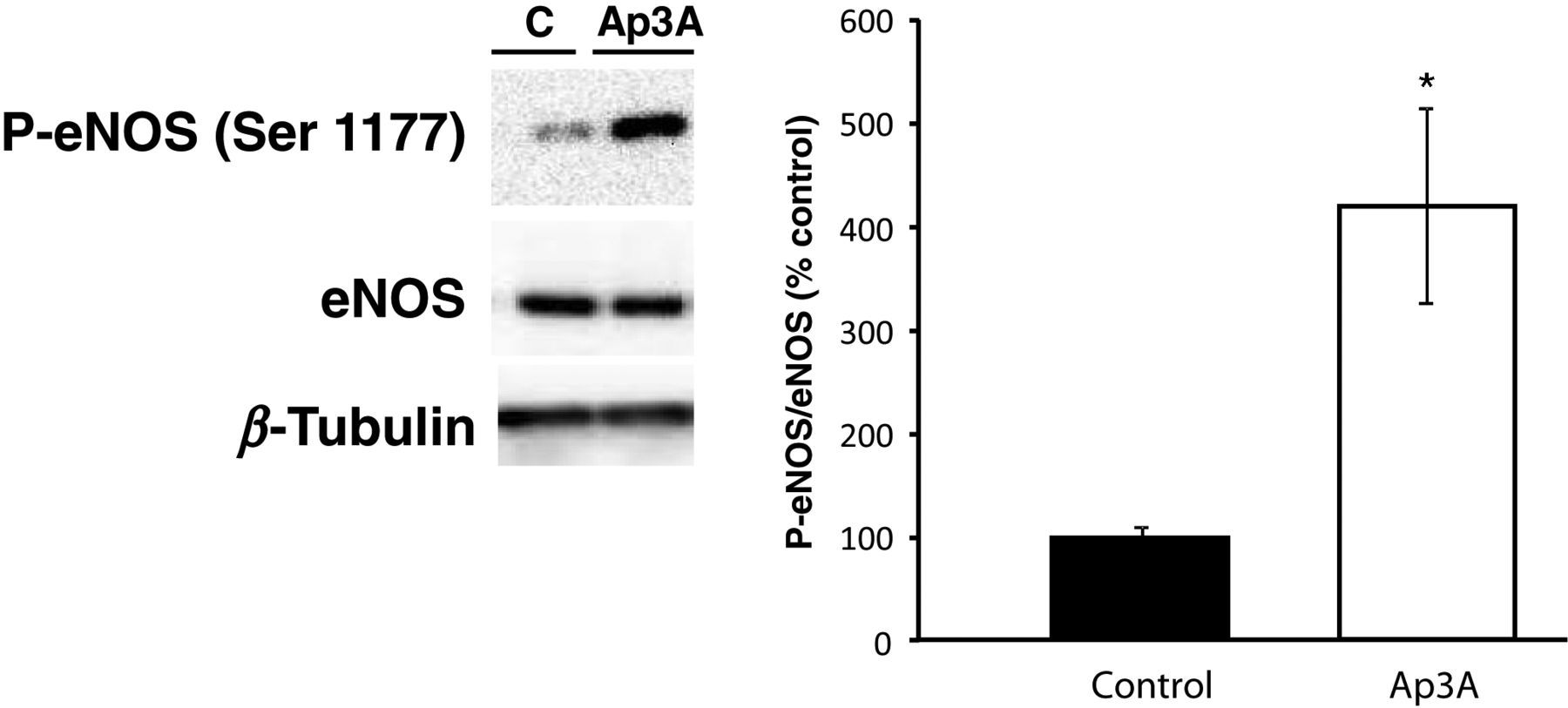
Discussion
Our results suggest that Ap3A promotes coronary vasodilation and that this effect is impaired after ischemia–reperfusion. The vasodilatatory effect of Ap3A in normal conditions was similar to that described for Ap4A in the canine heart, 14 for Ap3A, Ap4A, Ap5A and Ap6A in the guinea pig heart, 12 and for Ap4A and Ap5A in pig coronary arteries 13 and rabbit coronary circulation. 11 We previously demonstrated that Ap5A in control conditions induces coronary vasoconstriction followed by vasodilation. 18 By contrast, Ap4A induces vasodilation in control hearts but triggers a transient vasoconstrictor response after inhibition of purinergic P2y receptors, 17 while Ap3A induced vasodilation only (present study). Taken together, these findings suggest that diadenosines with a longer phosphate chain induce vasoconstriction, while those with a shorter chain elicit vasodilatator responses alone. 22
Several potential mechanisms may underlie the effects of ApnAs in blood vessels. First, ApnAs may activate P2 purinergic receptors, which mediate vasoconstriction and vasodilation via P2x and P2y, respectively. Purinergic P2y receptors have been proposed to mediate the vasodilatory effects of Ap4A in mesenteric arteries, both in humans
23
and rabbits,
24
that of Ap3A in rat mesenteric arteries,
25
and that of Ap5A and Ap4A in rat coronary arteries.
18,19
NO is also implicated in the APnA response, although its role may vary between animal species. For example, NO is involved in Ap4A-mediated coronary vasodilation in dogs,
14
but not in the effects of Ap3A, Ap4A, Ap5A or Ap6A on coronary circulation in guinea-pigs.
12
Moreover, NO is implicated in the vasodilatory effects of Ap4A but not Ap3A in rabbit coronary circulation.
11
Potassium channels may also be involved in the effects of Ap4A and Ap5A. Both drugs inhibit KATP potassium channels,
26
which may contribute to its vasoconstrictor effect, whereas KATP potassium channels in pig coronary arteries
27
are activated by Ap4A. The present findings suggest that all three mechanisms are involved in Ap3A-induced vasodilation of rat coronary circulation, as this effect was attenuated by reactive blue 2, MRS2179,
Ap3A-induced coronary relaxation was attenuated after ischemia–reperfusion and indeed, ischemia–reperfusion attenuates the vasodilatory effects of Ap5A 18 and Ap4A. 19 Moreover, ischemia–reperfusion impairs both NO-dependent 28 and KATP potassium channel-dependent relaxation 29 in the perfused rat heart, suggesting that impairment of these mechanisms is involved in the decreased vasodilatory response to Ap3A observed following ischemia–reperfusion. Functional impairment of P2y receptors may also contribute to this effect, as previously described for Ap4A 18 and Ap5A. 19
Our findings indicate that the no-reflow phenomenon, whereby coronary blood flow does not recover after ischemia–reperfusion, may be related to the impaired vasodilatory effects of Ap3A following ischemia–reperfusion. In agreement with previous findings, 18,19 coronary vasodilation in response to a APnA was disrupted after ischemia–reperfusion, an effect that may contribute to the reduced coronary blood flow in this condition.
In summary, the present findings demonstrate that ischemia–reperfusion impairs coronary vasodilation induced by Ap3A. Decreased purinergic P2y receptor function, reduced activation of KATP potassium channels and/or reduced release of NO release in coronary blood vessels contribute to this effect, which may be implicated in the pathophysiology of ischemia–reperfusion.
Footnotes
ACKNOWLEDGEMENTS
We are indebted to María Ester Martínez and Hortensia Fernández-Lomana for their invaluable technical assistance. This was financed by Fondo de Investigaciones Sanitarias (PS09/00394) and the Fundación Mutua Madrileña (AP57242009).