
Abstract
Hemozoin production makes it possible for intraerythrocytic malaria parasites to digest massive quantities of hemoglobin but still avoid potential ferriprotoporphyrin IX (FP) toxicity, which they cannot decompose further. Some antimalarial drugs, such as chloroquine, work by inhibiting this production, forcing the parasite to starve to death. As part of the efforts to identify possible biological mechanisms of FP polymerization, we have used normal human erythrocyte membranes as a model, to promote β-hematin (β-h) synthesis. Hemin in 35% aqueous dimethyl sulfoxide (DMSO) was reacted with isolated erythrocyte membranes and incubated overnight in sodium acetate buffer, pH 4.8, at 41°C. Infrared spectroscopy and electron microscopy showed that β-h was produced. Hemin in 10% was less effective as the substrate than when it was in 35% DMSO. A high malarial temperature seemed to be necessary, because FP polymerization was less at 37°C than at 41°C. Production was partially inhibited by chloroquine. These observations are of interest because other investigators have reported that membrane lipids mediated FP polymerization, but whole membranes were ineffective. On the other hand, our hypothesis is that the transport vesicles (TV) in malaria parasites could provide the receptor for FP and the lipids that promote hemozoin formation. Erythrocyte membranes may not be directly involved, but Plasmodium species transport hemoglobin in membrane-bound TV into food vacuoles, where hemoglobin catabolism is completed and hemozoin crystals are stored.
Introduction
Malaria remains a major parasitic disease of worldwide concern. Strains of the two Plasmodium species, P. falciparum and P. vivax, which cause most of the diseases and deaths in humans, have developed resistance against chloroquine, a drug that was once highly effective. 1 Attempts to understand how the drug works and the mechanism of resistance are currently focusing on how the parasite detoxifies the huge quantity of ferriprotoporphyrin IX (FP) that it liberates from hemoglobin during metabolism. The main process of FP detoxification in intraerythrocytic plasmodia involves formation of hemozoin, which happens largely in the food vacuole (FV), an acidic organelle of the parasite. 2,3 It is believed that without conversion to hemozoin, the potentially toxic FP could kill the parasites. Hemozoin is comparable to β-hematin (β-h), which can be readily synthesized in vitro, without the parasite, by heating FP at 60–80°C in acidic microenvironment, 3–5 or by other non-physiological methods in the absence of biological catalysts. 6,7 On the other hand, it is suspected that polymerization of FP at physiological temperatures in malaria infection might require some biomolecular catalysts. 8,9
Investigations were at one point focused on histidine-rich proteins, but this has been found to be limited to some P. falciparum strains. 10–12 Also, a heme detoxification protein that promotes FP polymerization has recently been described, 13 perhaps reviving the hemepolymerase hypothesis once propounded by Slater and Cerami. 14 Lipids have attracted considerable attention for some years as a potential promoter of hemozoin synthesis, 15–18 but investigators are wondering what the source of such lipids might be, having so far been unsuccessful in polymerizing FP with normal erythrocyte membranes. 16 One of us has previously suggested that malaria parasites use the membrane fragments released from hemoglobin transport vesicles (TV) in FV to initiate hemozoin formation. 19 The experiment on which that suggestion was based was constrained by the observation that normal erythrocyte membranes required prior activation with sodium hydroxide or acids before they could substantially promote hematin polymerization. 19 In the present study, we have used characterized hemin in aqueous dimethyl sulfoxide (DMSO) as the substrate for polymerization, without prior activation of the membranes with base or acid.
Among the factors that have not yet received sufficient attention is the possible state of free FP molecules in malaria parasites prior to incorporation into β-h. Here, we have used normal human erythrocytes, as a model, to investigate the reaction of monomeric and dimeric FP with membranes at conditions comparable to those in TV and FV of intraerythrocytic malaria parasites.
Materials and methods
Preparation of membranes
Blood was donated by healthy adult men and women with informed consent. It was collected in tubes containing the anticoagulant ethylenediaminetetraacetic acid. The blood samples were centrifuged for 10 min at room temperature and 3000 rpm, after which the supernatant plasma and white blood cells were discarded. The erythrocyte pellet was washed twice with 10 mL of cold saline solution, with centrifugation each time at room temperature and 3000 rpm. A suspension of the washed cells was prepared in normal saline solution to contain 1 × 109 erythrocytes/mL. Aliquots, each containing 2 mL of the cell suspension, were prepared in high-speed Oak Ridge tubes (Nalge Nunc International, Rochester, NY, USA) with screw caps. The aliquots were centrifuged cold for 30 min at 19,000 rpm. The supernatants were carefully removed and 10 mL of cold 5 mmol/L sodium phosphate solution, pH 7.6, was added to each tube. They were shaken vigorously by hand and kept on ice for 10 min to hemolyze. The tubes were centrifuged for 60 min at 19,000 rpm, after which the supernatants were discarded and the membrane pellets were washed twice by suspending them each time in 10 mL of cold 20 mmol/L Tris-buffered solution, pH 7.2, and centrifuging for 60 min at 19,000 rpm. The final pellet of erythrocyte membranes looked whitish to the naked eyes, indicating effective removal of contaminating hemoglobin. The tubes were stored frozen until required, usually within two weeks.
Use of human blood for this research project was approved by the Ethics and Research Committee of our University.
Substrate hemin solution and production of membrane-FP complex
A stock solution of bovine hemin, H9039, ≥90% pure (Sigma-Aldrich, St Louis, MO, USA), was prepared in concentrated DMSO, ≥99.9% pure (Merck, Darmstadt, Germany), and from it, suspensions containing a total of 2 × 109 of erythrocyte membranes in distilled water, 860 nmoles of hemin and 35% or 10% DMSO in a total volume 0.8 mL were prepared. The Oak Ridge tubes containing the suspensions were incubated for two hours at 37°C in a water bath shaker operating at moderate speed. Control samples contained the relevant solutions but without membranes. After the two hours of incubation at 37°C, 4 mL of 0.5 mol/L sodium acetate buffer, pH 4.57, was added to each tube and the suspension was mixed thoroughly. It was determined that this final suspension had pH 4.8. The tubes were then incubated further at 37 or 41°C for an additional 20 h (overnight). By increasing the total volume of the suspensions to 4.8 mL, the concentrations of DMSO in the originally 35% and 10% were dropped to approximately 5.8% and 1.7%, respectively, prior to the 20 h of incubation.
Extraction of total membrane lipids
The chloroform/methanol delipidation technic described by Di Girolamo et al. 20 was used to separate lipids from the other membrane components. The following chemicals were added into Eppendorf tubes (Eppendorf International, Pennsauken, NJ, USA) containing the membranes of 2 × 109 normal erythrocytes: 0.4 mL methanol, 0.1 mL chloroform and 0.3 mL distilled water. The suspension was thoroughly mixed and centrifuged in a microfuge for one minute. The water phase was removed and 0.4 mL methanol was added. After mixing thoroughly, it was centrifuged for two minutes. The supernatant was collected in a separate tube. The delipidated membrane pellet was air-dried at room temperature for 60 min and kept overnight. Similarly, the tube containing the methanol supernatant was left open to air-dry overnight at room temperature. Both samples were then used for FP polymerization together with tubes containing intact membranes.
Inhibition of membrane-FP complex formation by chloroquine
This part of the experiment was done just like the membrane-FP complex (MFPC) formation procedure described above, except that 50 μmol/L of chloroquine was added to the suspension right from the beginning.
FP aggregation at 41 and 60°C without membranes
In each of the tests with membranes, a membrane-free control was included. In addition, experiments were set up exactly as described for membranes, but without membrane, and were heated at 60°C for 20 h. The aggregates formed were purified with sodium dodecyl sulfate (SDS), NaHCO3 pH 9.1 or undiluted DMSO as described below.
MFPC purification with SDS
After a 20-h incubation of the different membrane-FP mixtures described above, the tubes were centrifuged at 19,000 rpm for 60 min. The pellet was suspended in 10 mL of 2.5% SDS buffer, pH 7.8, by vortexing, after which it was left at room temperature for two hours, with occasional shaking by hand and viortex, before centrifuging again at 19,000 rpm for 60 min. The pellet was suspended for the second time in 10 mL of 2.5% SDS buffer, pH 7.8, and then left at room temperature overnight to dissolve unincorporated FP and solubilize membrane proteins. The tubes were centrifuged at 19,000 rpm for 60 min and the supernatants were carefully discarded. After this purification, the aggregate formed with FP in 35% DMSO was code- named MFPC35D, and that formed with FP in 10% DMSO became MFPC10D. It should be pointed out that those DMSO concentrations remained only for the two hours prior to addition of 4 mL of sodium acetate buffer, after which the DMSO concentrations dropped to approximately 5.8% and 1.7%, respectively. In contrast, the total FP concentration was kept unchanged at 860 nmoles in all samples.
Additional purification of MFPC with NaHCO3 or undiluted DMSO
After the two-step extractions with SDS, the pellet was suspended either in 10 mL of 100 mmol/L NaHCO3, pH 9.1 or 10 mL of concentrated DMSO. The suspensions were kept at room temperature for 60 min, with occasional shaking, before they were centrifuged for 60 min at 19,000 rpm to recover the MFPC pellets.
Treatment of MFPC with proteinase K
Lyophilized proteinase K, together with the buffer for preparing the solution, was obtained from MACHEREY-NAGEL, Dueren, Germany. Fifty microliters of the enzyme solution was pipetted into the tubes containing MFPC and the suspension was mixed using a vortex. The tubes were incubated at 70°C for 3.5 h, followed by 10-min centrifugation in a microfuge. The supernatants were discarded and the pellets were washed thrice, using 1 mL of distilled water and centrifuging for 10 min each time. The pellets were then subjected to different analysis for β-h.
Estimation of FP concentration
The FP content of the MFPC before and after treatment with proteinase K, as well as all other FP concentrations, were calculated as described previously, 19 including solubilization in NaOH and 2.5% SDS buffer and using electronic absorbance at 400 nm wavelength.
Scanning electron microscopy
Both the proteinase K-treated and untreated MFPC extracts were fixed with 3% glutaraldehyde for 60 min and postfixed in aqueous osmium tetroxide for 60 min. They were washed and spread on coverslips and air-dried. The coverslips were mounted onto gold-coated stubs, using a sputter coater (Balzers SCD 050; Optics Balzers AG, Neugruet, Liechtenstein). A CarryScope JCM-5700 Mobile SEM instrument (Jeol USA Inc., Peabody, MA, USA) was used to scan for β-h crystals.
Transmission electron microscopy
MFPC samples were fixed with 3% glutaraldehyde and postfixed in 1% osmium tetraoxide for 60 min each. The pellets were embedded in 2% agarose and made into cubes. The agarose block was dehydrated with increasing concentrations of acetone and, after infiltration, embedded in fresh resin in a bead capsule. Hundred-nanometer thin sections, prepared and stained with uranyl acetate and lead citrate solutions, were taken with an ultramicrotome (RMC MT 7, Jeol USA, Inc.) and examined under a JEOL 1200 transmission electron microscope. The pictures were recorded using a computer connected to the microscope.
Hemolysis
Approximately 2.5 × 108 of washed plasma-free erythrocytes were suspended in 5 mL of Tris-buffered saline (TBS), pH 7.4, without or with chloroquine. A calculated volume of FP in undiluted DMSO was added to the erythrocyte suspension in Tris buffer such that the final proportion of DMSO in the suspension was 35% or 10%. The suspensions were incubated for two hours at 37°C in a water bath shaker operated at a moderate speed. After incubation, the hemolysates were centrifuged for 30 min at 3000 rpm before measuring absorbance of the supernatants at 540 nm wavelength. Percentage hemolysis was calculated relative to reference samples that were prepared together with each experiment and consisted of the mean absorbance of three hemolysates, each prepared by using 1 mL of 1% saponin in TBS to completely hemolyze 2.5 × 108 erythrocytes before adjusting the final volume to 5 mL with TBS. In the experiments testing the toxicity of DMSO, neither FP nor chloroquine were included in the erythrocyte suspension containing the required DMSO concentration.
Statistical analysis
Mean ± standard error, as well as t-test to determine P values, was done using GraphPad software (La Jolla, CA, USA).
Results
Spectral characterization of aqueous FP in DMSO
Ultraviolet/visible scans between 300 and 700 nm wavelength showed that hemin in undiluted (≥99.9%) DMSO had a tall Soret band at 402–405 nm, with other small peaks at approximately 500 and 625 nm wavelengths. The absorbance at the Soret band decreased with increasing dilution with distilled water. In a representative experiment (Figure 1), the absorbance of 10 μmol/L of FP in undiluted DMSO (Figure 1a) was 1.83 at 405, but decreased to 1.03 at 400 in 35% DMSO (Figure 1b). FP in 10% DMSO gave two peaks at the Soret band with absorbance 0.44 at 395 and 0.41 at 355 nm wavelength (Figure 1c). For comparison with FP in 10% DMSO, 10 μmol/L of FP in NaOH had absorbance 0.44 at 355 nm wavelength (Figure 1d).
Ultraviolet/visible spectrophotometric scans of hemin in dimethyl sulfoxide (DMSO) and NaOH. Stock solution of hemin was prepared in ≥99.9% DMSO (a) before diluting with distilled water to 35% (b) and 10% (c). However, all the solutions, including scan (d) which was prepared in NaOH, contained 10 μmol/L hemin. Maximum Soret band absorbance for a—d was at 405, 400, 395 and 355 nm wavelength, respectively. Note that ferriprotoporphyrin (FP) in 10% DMSO has a peak at 355 nm, in addition to that at 395 nm. The absorbance values were 1.83 at 405 nm (a), 1.03 at 400 nm (b) 0.44 at 395 nm and 0.41at 355 nm (c). For comparison, 10 μmol/L FP in NaOH also had 0.44 absorbance, but at 355 nm wavelength (d). (A color version of this figure is available in the online journal)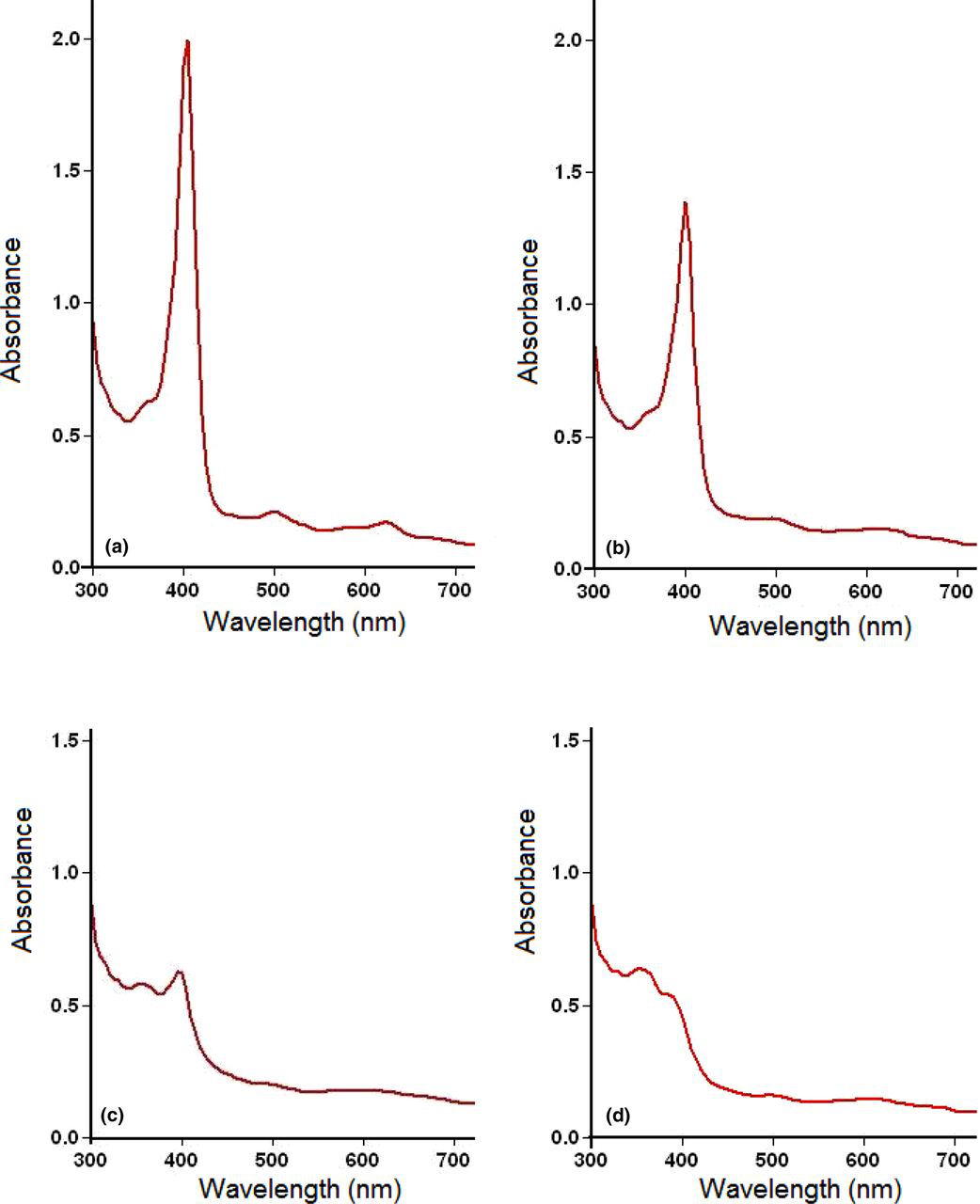
Effect of FP in aqueous DMSO on membranes at physiological alkaline pH
Toxicity of aqueous DMSO with and without hemin
DMSO, dimethyl sulfoxide
*Calculated by subtracting a from b
Effect of membranes on FP in acidic solution
Overnight incubation of FP and membrane mixture in sodium acetate buffer, pH 4.8, at 37 or 41°C, followed by purification with SDS, NaHCO3 or concentrated DMSO, produced an aggregate which we have referred to here as MFPC. Treatment with NaHCO3 or concentrated DMSO was intended to remove from MFPC any unincorporated FP aggregate that was not solubilized by SDS.
Repeated experiments showed that the quantity of MFPC synthesized was consistently lower when the reaction took place at 37°C than at 41°C. The yield shown in Figure 2a was calculated from MFPC that was purified with SDS and NaHCO3. Figure 2b shows that further purification of SDS-extracted MFPC with concentrated DMSO was more effective than with NaHCO3. Comparing the effect of chloroquine on membrane-mediated FP polymerization (Figure 2c), we found that there was a statistically significant inhibition (P < 0.05). Figure 2d shows that after lipid extraction from membrane, using the chloroform–methanol method, both the lipid fraction and the delipidated fraction still had FP polymerizing activity. The product shown in Figure 2d was purified with concentrated DMSO after double purification with 2.5% SDS. Except where 37°C is indicated, the polymerization results presented in these figures were obtained at 41°C. The MFPC in Figures 2a and c were purified with SDS and NaHCO3.
Quantity of ferriprotoporphyrin (FP) in membrane-FP complex (MFPC) under different conditions. The reaction in (a) was incubated overnight at either 37 or 41°C and MFPC was extracted with sodium dodecyl sulfate and NaHCO3, pH 9.1. Experiments summarized in Figure 2b tested for the effectiveness of NaHCO3 and DMSO in removing unpolymerized FP from MFPC. The inhibitory effect of 50 μmol/L chloroquine on MFPC production is shown in (c), whereas (d) shows that both delipidated membrane and the total lipid from the membrane promoted MFPC formation. β-h, β-hematin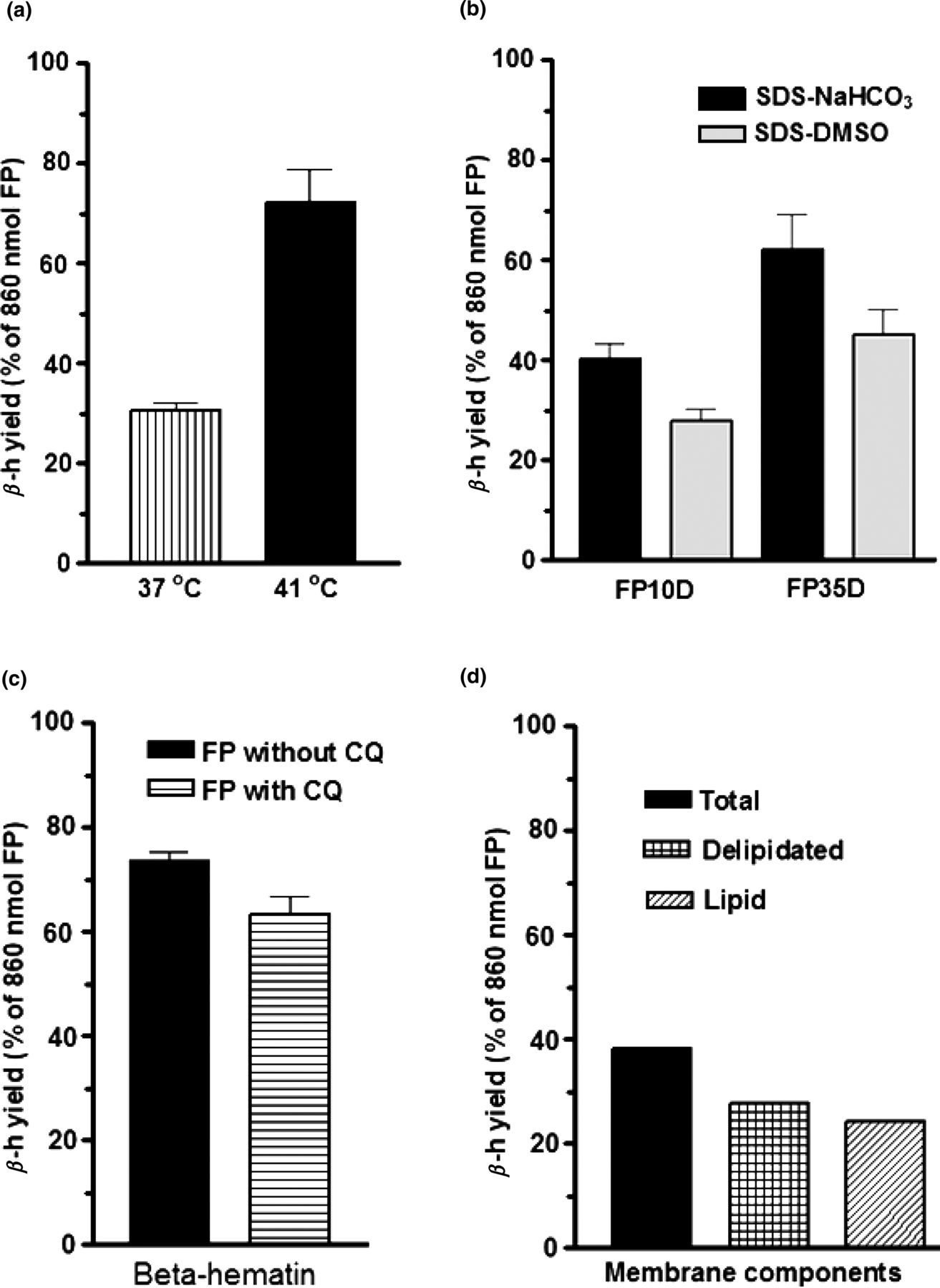
Fourier transform infrared spectroscopy
Figures 3a and b show Fourier transform infrared spectroscopy (FTIR) scans of MFPC before and after treatment with proteinase K. It is evident that there were proteinase K-susceptible functional groups that were interfering with β-h absorbance at 1662 cm−1. On the other hand, the peak at 1209 remained the same before and after proteinase K treatment. Slater et al.
3
have indicated that infrared absorbance around this wavenumber could be assigned to an axial carboxylate ligand.
Fourier transform infrared spectroscopy spectra of membrane-FP complex (MFKC) before and after proteinase K. Before treatment with the enzyme, the peak at 1209, but not that at 1640 cm−1, was correct for β-hematin (β-h) (a). The correct peaks for β-h (b) were obtained after treatment of MFPC with proteinase K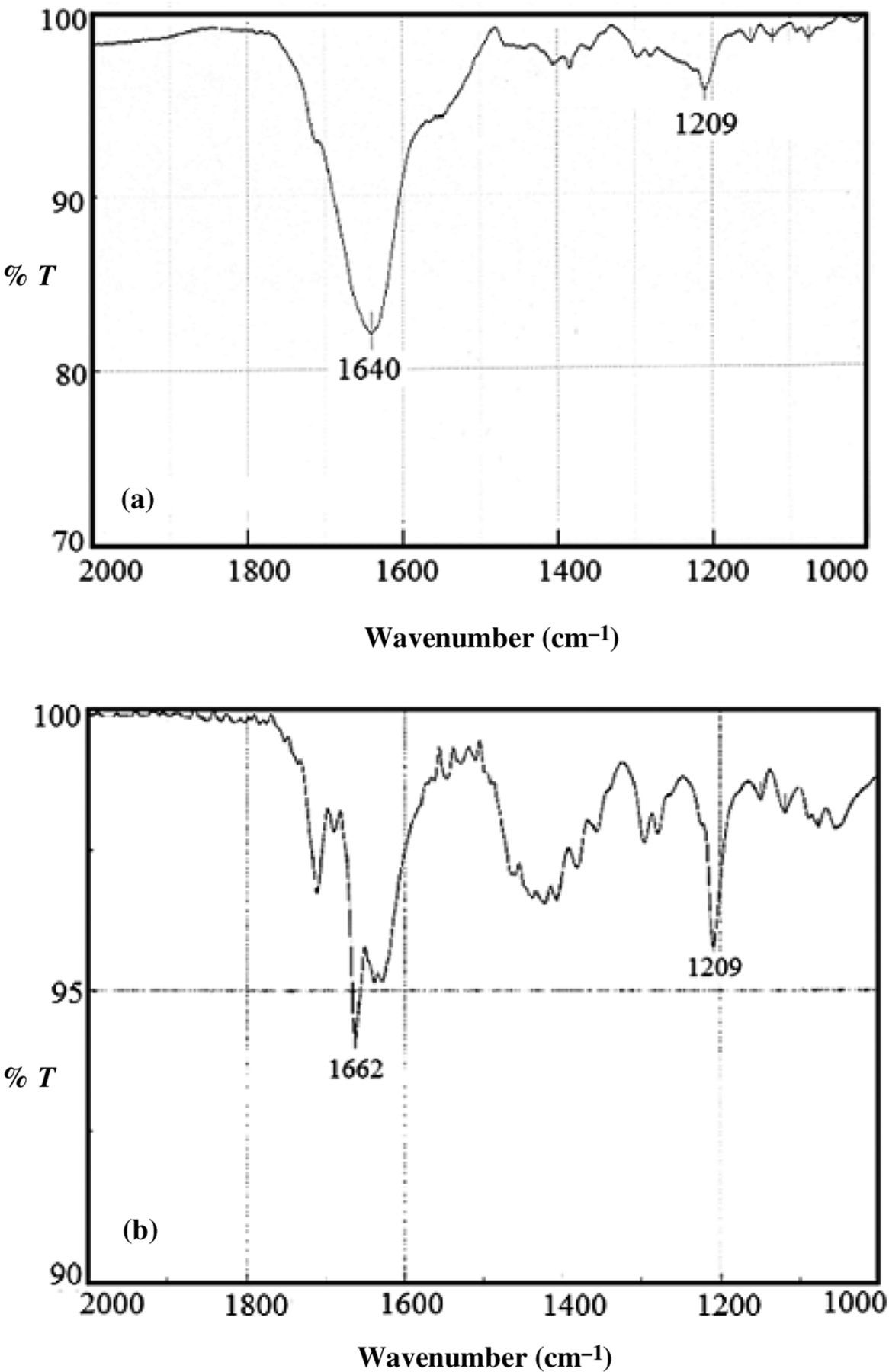
FTIR also gave some semi-quantitative information about MFPC by showing less intense absorbance at 1209 and 1662 cm−1 with FP10D than FP35D substrate (compare Figure 4a with b). The absorbance shown in the two figures were obtained when the MFPC products in two tubes of each substrate type were pooled. Comparing Figure 3b with 4b, the quantitative difference is also evident; Figure 3b was obtained with one tube of FP35D whereas Figure 4b was obtained with two tubes of FP35D pooled. Each tube contained the β-h product of 2 × 109 membranes and 860 nmoles of FP in 35% DMSO.
Fourier transform infrared spectroscopy (FITR) comparison of the effectiveness of FP10D and FP35D as substrates for membrane-mediated β-hematin (β-h) synthesis. Data summarized in (a) were obtained with FP10D, whereas those summerized in (b) were obtained with FP35D. Both of the samples were treated with proteinase K. In each case, two tubes containing the product were pooled before the FTIR shown here; in other words, the membrane-FP complex used in Figure 4b was double that of Figure 3b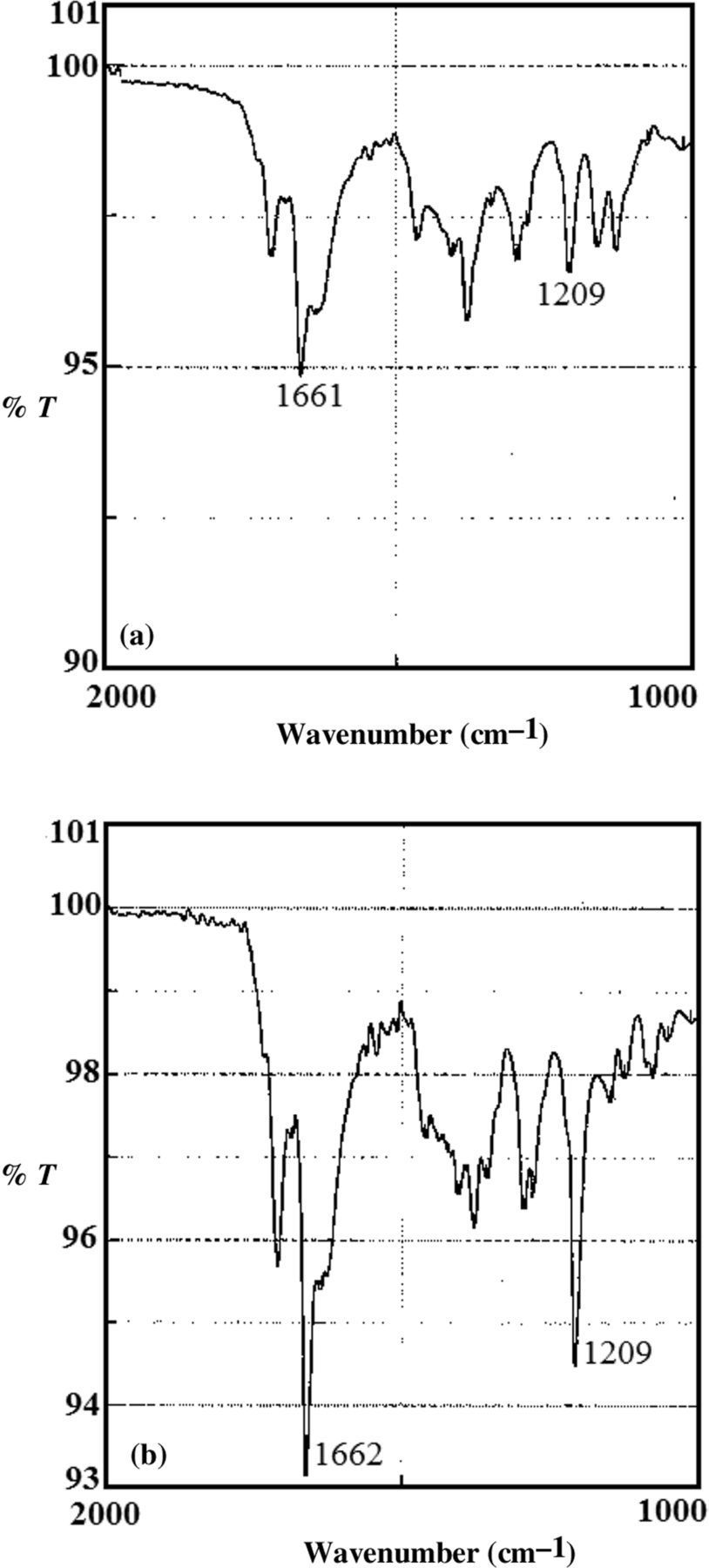
Scanning and transmission electron microscopy
Two photographs of scanning electron microscopy are shown in Figures 5a and b. As can be seen in Figure 5a, which shows MFPC before treatment with proteinase K, the enzyme treatment (Figure 5b) which required incubation at 70°C, played no role in the formation of β-h crystals. Limited measurement of the sizes showed that some of them were between 437 and 586 nm long and 127 and 143 nm wide.
Scanning electron micrographs of membrane-FP complex before (a) and after (b) treatment with proteinase K. Some of the β-hematin crystals ranged between 586 and 439 nm in length and 127 and 145 nm in diameter. (A color version of this figure is available in the online journal)
Figure 6 shows transmission electron micrographs of β-h crystals synthesized using FP35D and erythrocyte membranes at 41°C for 20 h. The top panel of the figure shows MFPC that was purified with SDS and NaHCO3; it contained membrane debris (arrows) as well as β-h crystals (dark particles). Treatment with proteinase K apparently digested away the membrane debris (bottom panel). Aggregates of the crystals in circular arrangement were observed in both untreated and enzyme-treated MFPC preparations.
Transmission electron microscopy micrographs of β-hematin crystals synthesized with erythrocyte membranes as promoter. The membrane-FP complex (MFPC) was synthesized at 41°C, using FP35D as substrate. Top and bottom panels show the crystals before and after proteinase K treatment, respectively. Apparently, the membrane debris in MFPC before proteinase K treatment (top panel arrows) was digested away by the enzyme (bottom panel)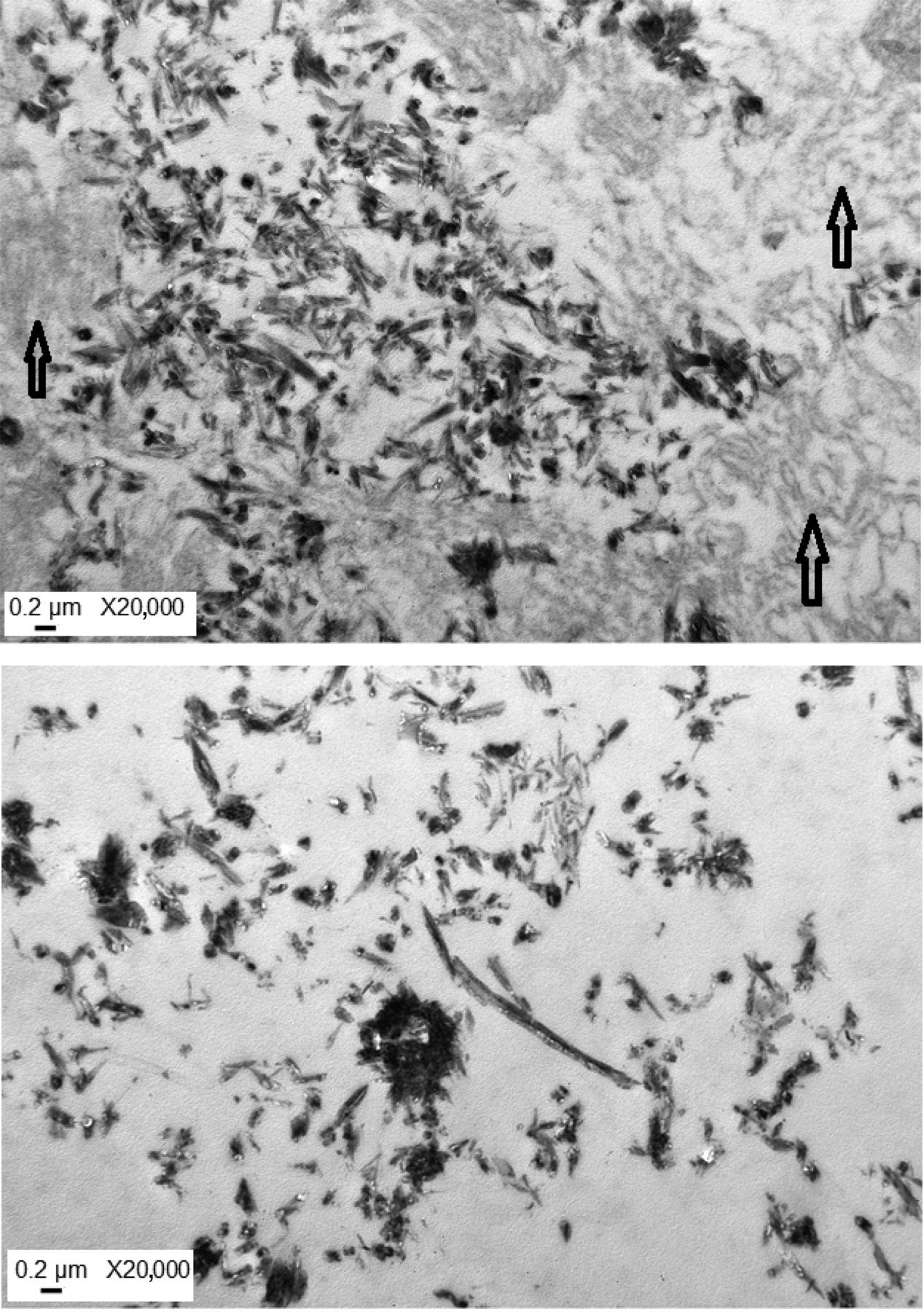
FP aggregation without membrane
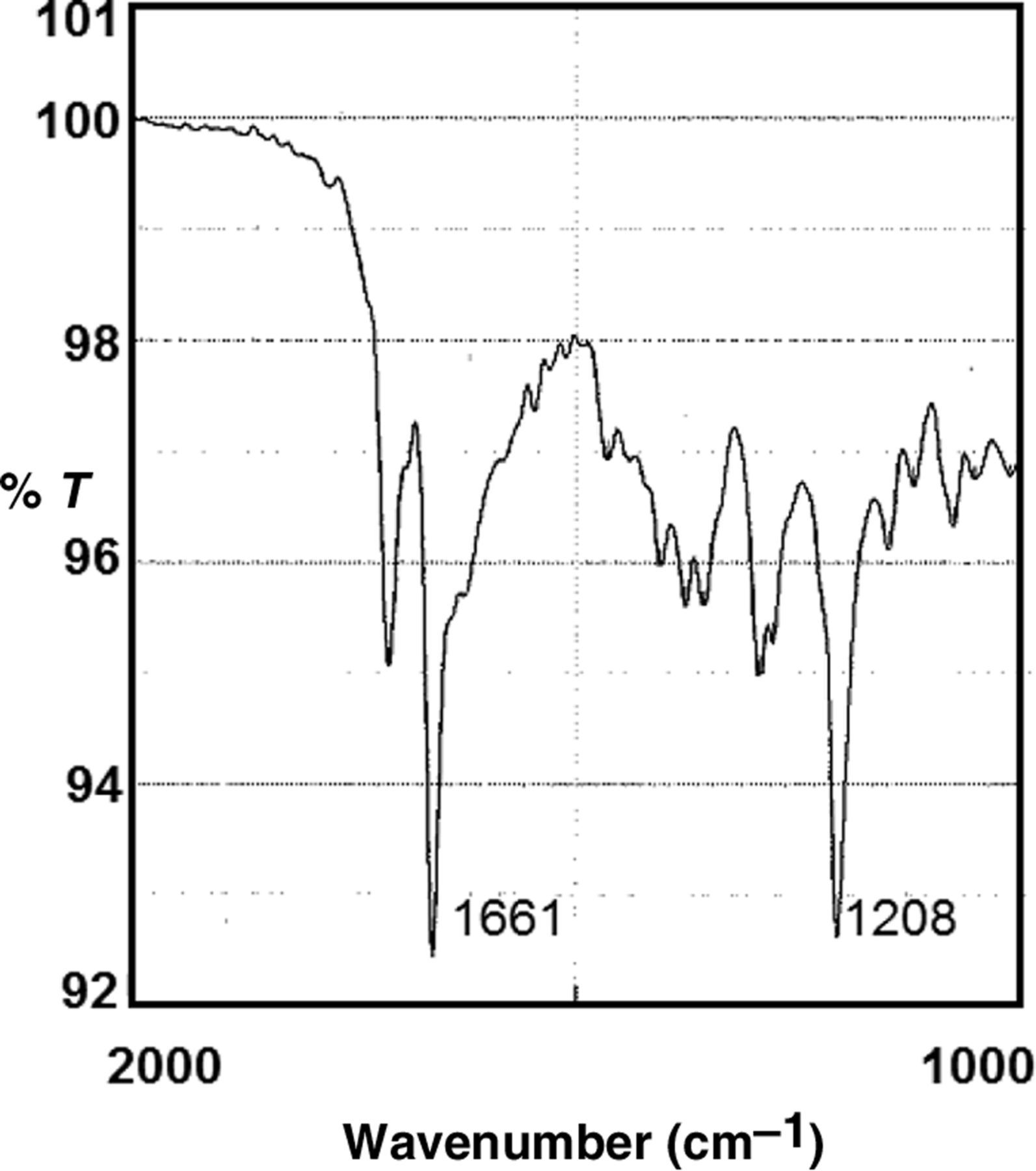
To show that membranes were the mediator of the β-h production described in this report, FP10D and FP35D were incubated at 41°C without membranes and extracted with SDS and NaHCO3, pH 9.1, or concentrated DMSO, exactly as was done with MFPC. As positive control for β-h production without a biological mediator, FP10D and FP35D alone in sodium acetate buffer, pH 4.8, were incubated for 20 h at 60°C and the aggregates were extracted using the same methods described for MFPC. It was found that at 41°C, about 2% of the 860 nmole substrate FP in FP10D and 14% of FP35D appeared resistant to the SDS-NaHCO3 extraction procedure (Figure 7, open bars). In contrast, incubation at 60°C produced relatively large quantities of the aggregate, apparently incorporating, on average, 38% of FP10D and 82% of FP35D (Figure 7, closed bars). Purification with SDS-DMSO decreased the aggregates formed at 41°C to approximately 0.6% and 5% using FP10D and FP35D, respectively (data not shown). After DMSO extraction of the aggregates produced at 60°C, approximately 29% and 50% of FP10D and FP35% were recovered, respectively, in β-h. The aggregate produced at 41°C was not sufficient for FTIR spectroscopy, but the product at 60°C had the β-h peaks at 1208 and 1661 wavenumber (Figure 8).
Ferriprotoporphyrin (FP) aggregation at high temperature without membranes. The substrates, FP10D and FP35D, were incubated at the indicated temperatures for 20 h. The aggregates were isolated and purified, using 2.5% sodium dodecyl sulfate solution in water, followed by NaHCO3 as described in the Materials and methods section. The shorter bar in the two temperatures were produced with FB10D, whereas the tall bar at each temperature was produced using FP35D Fourier transform infrared spectroscopy of β-hematin synthesized without membrane at 60°C. FP35D was used as substrate
Discussion
This study has, for the first time, presented infrared spectroscopy, as well as scanning and transmission electron microscopy of normal human erythrocyte membrane-mediated β-h synthesis. Previously, Fitch et al. 16 found that some membrane lipids purified from normal mouse erythrocytes had FP polymerization activity. The authors found that lipids extracted from Plasmodium berghei infected and uninfected mouse erythrocytes were qualitatively similar. However, when they used intact membrane ghosts, only the preparation from infected erythrocytes was active. Doing this type of experiment with parasitized erythrocytes is complicated by pre-existing hemozoin, which is difficult to remove completely from membranes, 19 and it is known that hemozoin can promote additional β-h formation. 3 Nevertheless, it was also found by Orjih 19 that there was no convincing polymerization of FP when hemin was dissolved in NaOH and incubated overnight with normal erythrocyte membranes in sodium acetate buffer, pH 5, at 37°C.
One of the decisive differences between the present work and the earlier report 19 is that well-characterized FP species were used here (Figure 1). Published electronic spectral analysis had clearly established that FP in 35 to 100% DMSO was in a monomeric state. 21 The chances of producing FP μ-oxo dimer increase as DMSO concentration decreases further. 22–24 Another new aspect in the present study is that by incubating at 41°C, we have mimicked malaria-induced rise at human body temperature 25 with success. It is obvious that not every person with malaria has a high temperature of 41°C; indeed, P. falciparum grows and produces hemozoin in vitro at 37°C. On the other hand, the temperature required for membrane-mediated FP polymerization in cell-free medium was clearly lower than the 60–80°C required for β-h synthesis in protein and lipid free systems. 3–5
The minimum aqueous DMSO concentration for maintaining FP at monomeric state was set at 40% by other investigators. 26 We chose 35% DMSO for our investigation after determining that 40% DMSO on its own caused, on average, 53% hemolysis of circa 2 × 108 human erythrocytes in two hours, compared with the 23% mean hemolysis caused by 35% DMSO (Table 1). Furthermore, in a preliminary FTIR spectroscopy experiment with MFPC that was not treated with proteinase K, β-h was detected when FP35D was subjected to polymerization, but not when FP40D was used as substrate (scans not shown). It could be that excess DMSO beyond 35% interferes with FP polymerization.
To test whether the superiority of FP35D over FP10D as substrate for membrane-mediated FP polymerization could be due to the toxic effect of DMSO on membranes, we have carried out β-h production at 60°C without membranes. From the data summarized in Figure 7, it was clear that the difference was also there. Another indication that DMSO toxicity on membranes may not explain the effectiveness of FP in 35% DMSO as a substrate for β-h synthesis can be deduced from the experiments showing that FP in 40% DMSO was even more damaging to membranes, as determined by hemolysis test, but did not produce FTIR demonstrable β-h. It is possible that FP35D had just the right proportion of monomeric FP and DMSO, so that any shift from this balance could interfere with FP polymerization. We are still investigating this possibility.
Regarding the relevance of the present study to hemozoin production in vivo, erythrocyte membrane-promoted FP polymerization could directly apply in the parasitic worm Schistosoma where the fluke ingests blood and digests it whole, although promotion by extracellular lipid droplets have been reported. 27 In contrast, the feeding mechanism of malaria parasites in erythrocytes involves formation of small erythrocyte-like organelles called TV, in which hemoglobin is delivered into FV. Like the host erythrocyte, TV is surrounded by membrane and contains the hemoglobin of the host cell. TV is initially surrounded by double membranes, but the outer one is lost during entry into FV. 27,28 Although there could be some chemical and antigenic differences between the membrane of the host cell and TV, both are made of lipid bilayer. At all times during transportation into FV, hemoglobin is in close contact with the TV inner membrane, which is derived from the parasitophorous vacuole membrane (PVM). It has been demonstrated that the lipid composition of PVM is similar to that of the erythrocyte plasma membrane. 29 We hypothesize that while being transported to FV, the hemoglobin in TV denatures, perhaps due to adverse conditions, and because enzymatic digestion of hemoglobin probably begins even before TV gets into FV. 30 Such precipitates, perhaps comparable to denatured hemoglobin or hemichrome in homozygous sickle cell (SS) and other erythrocytes types, 31–34 could expose FP molecules that could react with TV membrane. Chloroquine-accessible FP has been previously detected in SS erythrocytes, which was taken as indirect evidence of FP accumulation in erythrocytes containing denatured hemoglobin. 35,36
The state of hemoglobin during enzymatic digestion in malaria parasites is probably not yet completely defined, but it has been determined that some enzymes involved in intra-FV proteolysis are particularly effective on denatured hemoglobin. 37 Our work here with MFPC and proteinase K indicates that enzymatic digestion could continue even after FP has formed complexes with membrane components. Perhaps as proteins are being digested, freed membrane lipids could mix with FP aggregates and create a hydrophobic micro-environment, making it easier for the necessary chemical bonds to form between FP molecules. This scenario is consistent with the hypothesis put forward by Pisciotta et al. 38 on the possible role of neutral lipid nanospheres in hemozoin synthesis in malaria parasites. However, it is unclear whether lipids are the sole mediator of FP polymerization, because we found here that delipidated membrane was still effective (Figure 2d).
In conclusion, our primary objective to present clear evidence showing that normal human erythrocyte membranes can mediate production of synthetic β-h has been achieved. The two factors that have made this possible were (1) careful titration of the substrate hemin-DMSO mixture and (2) taking malaria-induced increase in body temperature into consideration. Furthermore, it was helpful to let the monomeric FP in DMSO gain access to the membranes first before being precipitated out with acidic sodium acetate buffer to initiate polymerization.
Footnotes
ACKNOWLEDGEMENTS
This work was supported by Kuwait University, Research Grant No. NM03/05. We thank Professor David Sullivan of Johns Hopkins University, USA, for FTIR analysis of our preliminary MFPC samples. The electron microscopy facilities at the Faculty of Medicine and Faculty of Sciences, Kuwait University, were used for the relevant examinations shown in this report. We also thank GRFP GS 01/05 Division at Faculty of Science, Kuwait University, for the FTIR scans.