
Abstract
Myocardial remodeling after ischemic infarction is characterized by collagen accumulation leading to replacement and interstitial fibrosis. Type I and III collagens are predominant components in cardiac fibrosis. Lysyl oxidase (LOX) facilitates the cross-linking of type I and III fibrils, resulting in the formation of stiff fibers and their subsequent tissue deposition. However, the matrix metalloproteinases (MMPs), a family of zinc-dependent enzymes, function in the degradation of the collagen components of extracellular matrix. Tissue inhibitors for MMPs (TIMPs) manipulate the action of MMPs. To understand the contribution of these molecules to cardiac fibrosis, we developed a rhesus monkey model to determine the changes in LOX, MMP1 and TIMP1 in relation to collagen deposition after myocardial ischemic infarction. Male rhesus monkeys were subjected to left anterior descending artery ligation along with sham-operated controls. Histological examination and immunochemistry were performed eight weeks after the ischemic injury. The results showed that both type I and III collagens were increased in the scar area and in the interstitium, and the ratio of type I/III collagens also increased in the scar area but not in the interstitium. The expression of LOX was up-regulated, but the expression of MMP1 was down-regulated in residual myocytes of the scar area and the border zone. The expression of TIMP1 was not changed. The data thus demonstrated that the collagen deposition in infarcted myocardium is correlated with an enhanced cross-linking capacity and a decreased degradation process.
Introduction
Myocardial infarction is a major cause of death and disability over the world. 1 This pathogenesis includes coronary artery occlusion, ischemic death of myocardial tissue and myocardial remodeling along with left ventricular dilation and decreased cardiac function. 2,3 Myocardial remodeling plays a critical role in overt heart failure, which is characterized by an accumulation of collagen in ventricles, leading to replacement and interstitial fibrosis. 4 Types I and III collagens are the predominant components of the collagen fibrils deposited in the heart following myocardial infarction. 5,6
Collagen synthesis and degradation are closely regulated by several factors, including matrix metalloproteinases (MMPs) and tissue inhibitors of matrix metalloproteinases (TIMPs). 7 MMPs are a family of zinc-dependent enzymes that can degrade components of the extracellular matrix (ECM). The MMPs family can be divided into several classes according to substrate specificity: the collagenases (e.g. MMP-1) that degrade fibrillar collagens including types I, II and III; the gelatinases (e.g. MMP-2 and MMP-9); the stromelysins (e.g. MMP-3); and the membranous type MMP (e.g. MT1-MMP). The activities of MMPs can be inhibited by TIMPs, a group of endogenous MMP inhibitors. Dysregulation of MMPs/TIMPs is associated with myocardial remodeling. 8,9
The enzyme lysyl oxidase (LOX) is a copper-dependent extracellular enzyme that catalyzes lysine-derived cross-links in collagen and elastin. LOX facilitates the cross-linking of collagen types I and III fibrils, leading to the formation of stiff collagen types I and III fibers and their subsequent tissue deposition. Several studies have suggested a strong association between organ fibrosis and increased LOX expression. 10,11 Five different, but closely related, LOX isoenzymes have been identified (LOX, LOXL-1, LOXL-2, LOXL-3 and LOXL-4), and LOX is the most abundant form in the heart. 12
The dynamic changes among these fibrogenesis and fibrolysis proteins determine the ultimate pathogenesis of cardiac fibrosis. The roles of these proteins in myocardial collagen deposition have been extensively studied in rodent models. However, the information generated from rodent studies cannot be directly extrapolated to humans. In this context, rhesus monkeys have been considered as the most valuable non-human primate animal models for experimental studies of human physiology and pathogenesis because of their close similarity to humans in many physiological aspects. 13 For the studies of cardiovascular diseases, especially heart disease, rhesus monkeys are important substitutes for humans. The heart of rhesus monkeys highly resembles that of humans. 14 Therefore, the rhesus monkey model of myocardial infarction would provide a better surrogate for the same condition in humans.
In the present study, the myocardial collagen content and composition (the ratio of type I/III) changes in the rhesus monkey model of chronic myocardial infarction were evaluated in association with the changes in LOX, MMP1 and TIMP1. The goal was to define the contribution of these proteins to cardiac fibrosis. The results showed that differentially localized up-regulation of LOX and down-regulation of MMP1 were associated with collagen deposition following myocardial infarction.
Materials and methods
Animal models
Male rhesus (Macaca mulatta) monkeys, aged 2–3 years old and weighing 4.5–6.0 kg, were obtained from Chengdu Ping-An Experimental Animal Breeding and Research Center, a Chinese government-accredited non-human primate center in Sichuan province. The animals were housed in individual cages and acclimatized to the laboratory condition for a period of at least one month. They received a conventional laboratory diet with free access to drinking water as approved by the Laboratory Animal Management Committee of Sichuan province. All animal procedures were approved by the Institutional Animal Care and Use Committee at the Sichuan University West China Hospital, following the guideline of the US National Institutes of Health.
Animal surgery preparation
Prior to the surgical procedure, all subjects received an intramuscular injection of 10 mg/kg ketamine and 0.2 mg/kg midazolam to induce sedation while they were maintained in a supine position on the operating table. Each subject was prepared surgically with a chronic indwelling venous catheter under sterile conditions.
Induction of myocardial ischemic infarction
A total of nine monkeys were randomly divided into two groups: ischemic (n = 4) or sham-operated control (n = 5). Standard non-invasive measurements including electrocardiography, cuff blood pressure, pulse oximetry and capnography were constantly monitored. Intra-arterial and intravenous catheters were used. Anesthesia was induced by intravenous infusion with fentanyl (10 μg/kg), midazolam (0.2 mg/kg), propofol (1 mg/kg) and vecuronium (0.1 mg/kg). Fentanyl and vecuronium were also intermittently infused in addition to isoflurane (1.0–2.5%) inhalation to maintain the anesthetic condition during the surgical procedure. All of the monkeys subjected to the surgical procedure were intubated and ventilated to achieve end-tidal CO2 between 35 and 40 mmHg.
The heart of monkeys was exposed via a sternotomy using rib shears. The left anterior descending artery (LAD) was ligated between the first and the second diagonal artery with 4-0 polyethylene sutures after repeated ischemia for two times (occlude the artery for 1 min and then reperfusion for 5 min). Dobutamine (3–5 μg kg−1 min−1) was infused after the LAD ligation to support the cardiac function if necessary. The pericardium and pleura were closed with 4-0 polyethylene sutures after the cardiac hemodynamics became stable. The sternum incision was closed with 10 silk sutures and the skin incision was closed with 1.0 silk sutures. The endotracheal tube was retracted after spontaneous breathing was restored. The sham-operated controls were subjected to the same surgical procedure with the exception of LAD ligation.
Preparation of tissue sections
On the 56th postoperative day, the animals were anesthetized with ketamine and euthanized by intravenous injection of potassium chloride. Their hearts were removed from the chest, and flushed by saline after stripping of the pericardium. The gross pathological observation was made, and then the hearts were fixed in 10% formalin. The atrium (the upper part of the mitral valve plane) was excised first, then the left ventricle was sectioned horizontally with equal thickness (4 mm) from the apex to the basal part of the left ventricle, and each slice was embedded in paraffin. 15 A serial of 4-μm sections were made from each block. One set of the sections was stained with hematoxylin and eosin, Masson's trichrome (MTC) and collagen-specific picrosirius red, and the other was used for immunohistochemistry.
Measurement of myocardial infarct size
MTC-stained sections were captured as digital images, and the following parameters of each image were measured using Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA): the epicardial infarct length (the length of the transmural infarct region), the endocardial infarct length (the length of endocardial infarct scar surface >50% of the whole thickness of myocardium) and the epicardial and the endocardial circumferences. The infarct size was calculated as ([sum of epicardial infarct lengths/sum of epicardial circumferences + sum of endocardial infarct lengths/sum of endocardial circumferences]/2)×100. 16
Measurement of collagens
Both scar replacement and interstitial collagens of picrosirius red-stained sections were imaged using a circularly polarized light microscope and the collagen volume of each type was measured by Image-Pro Plus 6.0. The collagen fiber color in picrosirius red-stained sections viewed under a polarized light microscope varies with the thickness: collagen I appears as yellow–red fibers, whereas collagen III appears as green fibers. 17 Collagen volume fraction (CVF) was determined as the ratio of connective tissue areas divided by the total tissue area within the same microscopic field. The ratio of type I collagen to type III was calculated for each field (the red area/the green area).
Immunohistochemical staining
An immunohistochemical streptavidin-peroxidase method was used to detect LOX, MMP-1 and TIMP-1. 18 Paraffin-embedded sections were de-paraffinized in a series of ethanol and xylenes. Sections were blocked with methanol containing 3% hydrogen peroxide (H2O2) for 10 min at room temperature, sequentially 10% goat normal serum for 30 min at room temperature and incubated with the primary antibody (rabbit polyclonal antibody against human LOX, 1:400; rabbit monoclonal antibody against human MMP1, 1:100; mouse monoclonal antibody against human TIMP1, 1:50; Abcam plc, Cambridge, UK) overnight at 4°C in a humidified chamber. After incubation, unbound antibodies were removed by three washing steps with phosphate-buffered saline for five minutes. Subsequently, the tissue sections were sequentially treated with biotinlabeled secondary antibody (Zhongshan Golden Bridge Inc., Beijing, China) and horseradish peroxidase-labeled streptavidin for 30 min at 37°C, respectively. A 3,3’-diaminobenzidine kit (Zhongshan Golden Bridge Inc.) was used to develop the positive reaction as a brown color. Finally, the tissue sections were counterstained by hematoxylin to visualize nuclei and coverslips were placed over them. Immunostaining of these proteins on the tissue sections was detected by a light microscope and the images were digitized. Under ×200 magnification, 10 visual fields were randomly observed, testing the average optical density of cells with positive expression (integrated optical density/area) for semi-quantitative statistical analysis. 19 Phosphate-buffered saline was used as the negative control.
Statistical analysis
Data are presented as mean ± SD. Student's t-test and one-way analysis of variance were used for statistical evaluation. SPSS (version 17.0; SPSS Inc., Chicago, IL, USA) software was used for the analysis. Statistical significance was assumed when P < 0.05.
Results
Histopathological changes and infarct size
The hearts of all animals were examined histologically. Absence of myocardial infarction was confirmed in all control animals. All animals subjected to LAD ligation for eight weeks showed left ventricle transmural infarction. As shown in Figure 1, infarcts were located in the left ventricle apex, low part of the left ventricular anterior wall and interventricular septum. The infarct size was 21.82 ± 1.73%. The infarct areas showed typical histopathological features, consisting mostly of dense collagens with sparse and mature fibrocytes. In addition, inflammatory cell infiltrations including a small number of macrophages, lymphocytes and plasma cells were identified in the infarct areas.
Histopathological observation of myocardial infarction on the 56th day after LAD ligation in rhesus monkeys. (a) Overview of the entire heart with apparent infarct areas identified (red arrow); (b) thin slices showing different sections of the heart with infarct areas including left ventricle apex, low portion of the interventricular septum, and some parts of the right ventricle (red arrows); (c) Masson-stained sections showing infarcted areas (red arrow). LAD, left anterior descending artery. (A colour version of this figure is available in the online journal)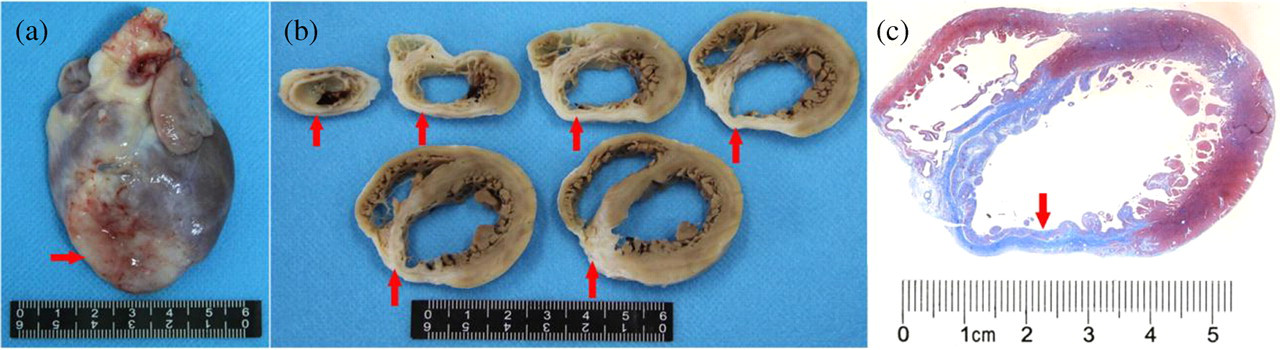
Collagen type composition
Under a polarized light microscope, the collagen fibers could be seen clearly in picrosirus red-stained sections in both the interstitium and the infarct area (Figure 2a). Compared with the ECM of the sham-operated group (ECM-sham), the interstitial CVF in the ECM of the myocardial infarction group (ECM-MI) increased significantly (Figure 2b). The ratio of collagen I/III significantly increased in the infarct scar (scar-MI), but did not change in the interstitium of the infarcted myocardium (ECM-MI) in comparison to that of the ECM of the sham-operated controls (ECM-sham) (Figure 2c).
Picrosirius red staining of collagens in myocardial infarct areas and the surrounding zones, and semi-quantitative analysis of collagen deposition. (a) The photomicrographs of left ventricular tissue sections stained with picrosirius red (×200). The enhanced deposition of collagen in ECM-MI and scar-MI compared with ECM-sham can be seen on the left panels. The polarization microphotography of picrosirius stained LV sections, green collagen fibers (type III collagen) and yellow–red collagen fibers (type I collagen) are shown in the right panels; (b) quantitative analysis of the collagen volume fraction (CVF) in ECM-sham and ECM-MI; (c) the ratio of type I/III collagen among ECM-sham, ECM-MI and scar-MI; *indicates P < 0.05. ECM, extracellular matrix; MI, myocardial infarction. (A colour version of this figure is available in the online journal)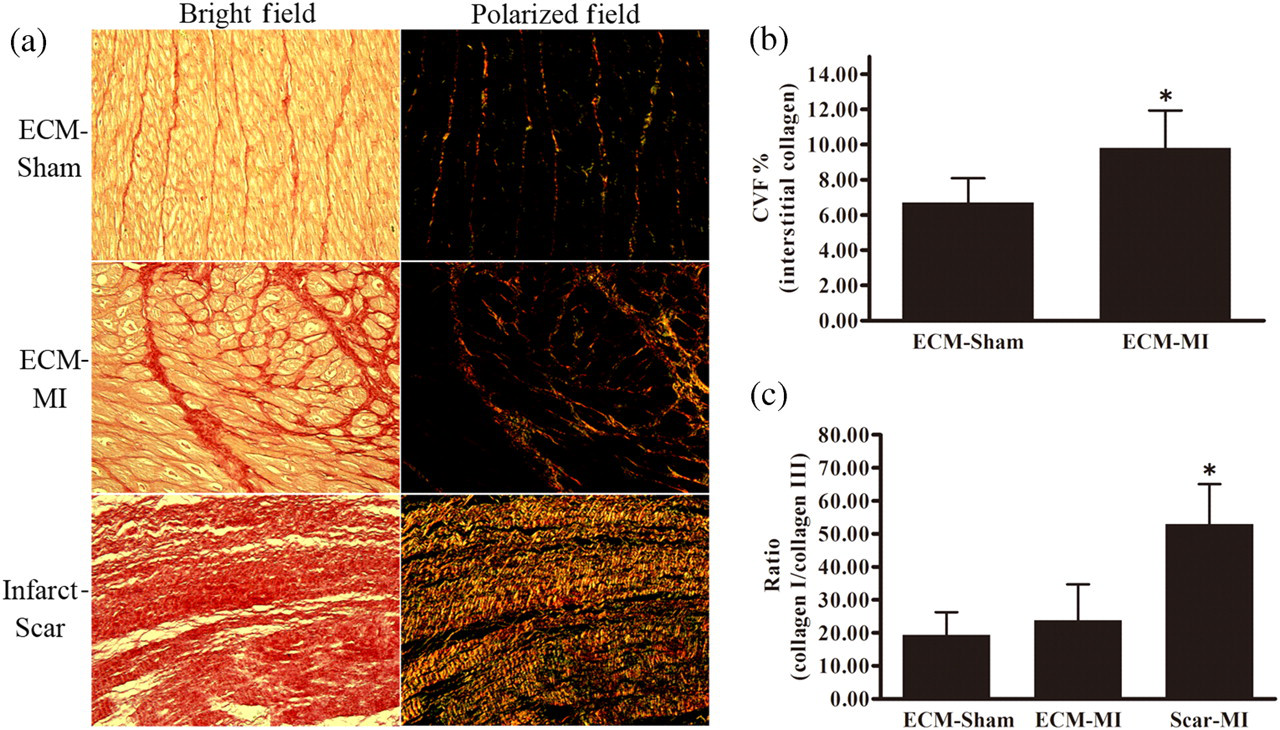
LOX, MMP1 and TIMP1 expression
The expression of LOX, MMP1 and TIMP1 was detected immunohistochemically, as shown in Figure 3. Myocardial LOX protein in the peri-infarct zone (PIZ) was significantly increased compared with the sham-operated controls (Sham), but far beyond the infarct zone (FIZ), LOX expression was only slightly increased (Figure 3b). Myocardial MMP1 protein in the PIZ was significantly decreased in the myocardial infarction group compared with the sham-operated controls, but only slightly decreased in the FIZ area (Figure 3c). Myocardial TIMP1 protein in both the PIZ and FIZ did not show significant changes in comparison with sham-operated controls.
Immunohistochemical detection of myocardial expression of lysyl oxidase (LOX), matrix metalloproteinase 1 (MMP1) and tissue inhibitor for MMP 1 (TIMP1). The photomicrographs of left ventricular tissue sections showing LOX, MMP1 and TIMP1 staining (3.3’-diaminobenzidine; ×200). Nuclei were stained blue by hematoxylin (a). Average optical density (AOD) value of LOX, MMP1 and TIMP1 (b–d). Data are presented as mean ± SD. *Indicates P < 0.05. (A colour version of this figure is available in the online journal)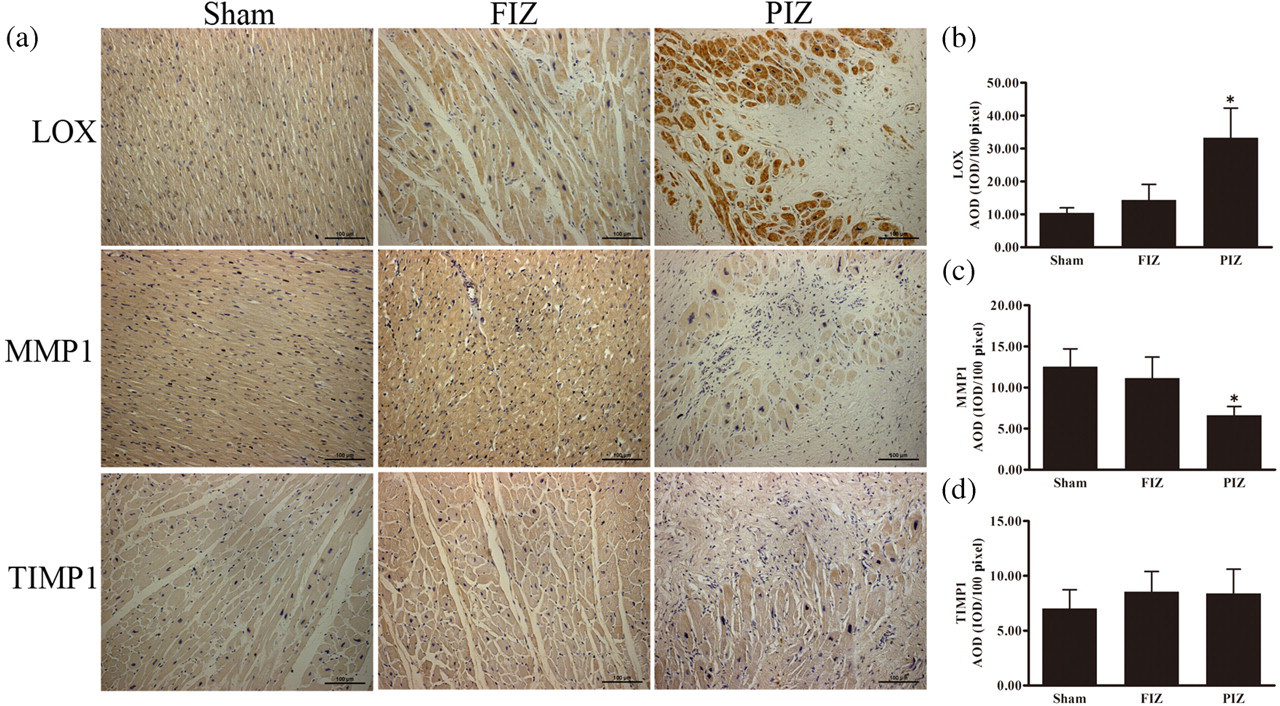
Discussion
In the present study, the non-human primate rhesus monkey model of myocardial ischemic infarction was used as a better surrogate for human disease condition. The analysis of infarcted hearts at eight weeks after LAD ligation revealed significant changes in collagen deposition and alterations in MMP1 and LOX in the infarct area, PIZ and far beyond the infarct area. These changes were in favor of the process of cardiac fibrosis. Scar replacement and interstitial fibrosis were evidenced by the increased deposition of collagen I and III in the myocardium subjected to LAD ligation. The increased collagen deposition was accompanied by decreased expression of MMP1 and increased expression of LOX, facilitating the cross-linking of the collagen fibrils and promoting the formation of stiff collagen fibers for deposition. This observation in the monkey model would be more closely extrapolated to the human disease condition.
Collagen deposition following myocardial infarction is a critical process of myocardial remodeling, which is mainly characterized by scar replacement and interstitial fibrosis. Types I and III collagens are the major components of cardiac fibrosis. Prior studies have shown that changes in collagen quantity and composition play an important role in the development of left ventricular dysfunction induced by myocardial infarction in animal models. The increase in collagen content has been observed in the remaining viable free wall and the ratio of types I and III collagen was found to be increased in the infarcted area. 20,21 The increases in the total content of collagens and the ratio of type I to type III collagens make important contributions to not only cardiac stiffness deterioration, but also the disruption of electrical and mechanical properties of the heart. 22
The increase in collagen production and deposition would activate the counteracting system, such as increased expression of MMPs, particularly MMP1 and MMP13, for the degradation of deposited collagen fibers. Another regulatory mechanism is TIMPs, which negatively regulate the activity of MMPs, leading to the prevention of fibrolysis by MMPs. The alteration of MMPs/TIMPs has been well recognized to be involved in myocardial remodeling. 23 The observation in the present study, that eight weeks after LAD ligation in the monkey model, the increased deposition of collagen fibers was associated with decreased expression of MMP1, and not an increase in the expression, was interesting. Human biopsy tissue sample analysis of cardiac fibrosis often showed increased levels of MMPs. 24,25 This apparent discrepancy provides an alternative insight into the pathogenesis of cardiac fibrosis. In comparison to human studies of cardiac fibrosis, the monkey model presented here would represent the early stage of myocardial fibrogenesis. The depressed expression of MMPs, rather than increased expression, would be the major problem during the early stage of the pathogenesis.
The other major contribution to cardiac fibrosis is the cross-linking of collagen fibrils. LOX is the critical enzyme responsible for cross-linking of collagens in the myocardium. LOX makes soluble collagen molecules into insoluble fibrous organization through the cross-linking reaction. 26,27 A remarkable up-regulation of LOX has been demonstrated in the myocardium after myocardial infarction. 28 Inhibition of LOX activity is beneficial because it limits the development of left ventricular stiffness in failing hearts, which results from excess collagen deposition at myocardial lesions. 29 The observation presented here recapitulated the clinical observation. The increased expression of LOX in the residual cardiac cells in the infarcted area would greatly facilitate the deposition of collagens in this area. This increase, along with the depressed expression of MMP1, made the pathogenesis of cardiac fibrosis feasible.
The increased ratio of type I/III collagen in the scar area was an important observation in the present study. This increase would make the deposited collagens stiffer due to the fact that the type I collagen is much thicker than the type III collagen. It was interesting to observe that the increase in the ratio of type I to type III collagens only occurred in the infarcted area, and not in the interstitium, although the total content of collagens was increased in both areas. Corresponding to this differential change, the increased expression of LOX occurred in the residual cells in the infarct area and the board zone of the scar area. Is LOX responsible for the increase in the ratio of type I to type III collagens in the scar area? This is an interesting question that should be answered in the future.
In the past several decades, most studies of cardiac remodeling postmyocardial infarction have focused on the changes of interstitial collagen. As for the infarct scar, studies have focused on how to increase its thickness to prevent cardiac rupture. However, the infarct scar may impair the normal contractile function of the myocardium and cause arrhythmia. 30 Recent studies have shown that increased infarct wall thickness was insufficient to prevent negative left ventricular remodeling and degradation of the infarct scar was also important for repairing cardiac tissue after myocardial infarction. 31,32 It has been challenged in the past few years that the adult mammalian heart is a terminally differentiated organ. 33–35 The infarct scar is a barrier for cardiomyocyte regeneration mainly because it impairs penetration of stem/progenitor cells into infarcted areas. 36 The present study demonstrated that modulating the level of LOX and MMP1 may help in developing methods promoting degradation of collagen in the scar and regeneration of cardiomyocytes after myocardial infarction.
In summary, the data obtained from the rhesus monkey model of myocardial infarction identified by the immunohistochemical method differentially localized up-regulation of LOX and down-regulation of MMP1 in the PIZ. This alteration was accompanied with increased deposition of collagens and increased ratio of type I to type III collagens in the infarcted area. Therefore, in the early stage of myocardial infarction, the molecular changes would be in the process of promoting collagen deposition.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Xiaohe Chen and Xiuqun Li for technical support. This work was supported by the Science Foundation of Sichuan Province (2009HH0016; 09ZQ026-081) and by US National Institutes of Health (HL63760 to YJK).