
Abstract
Enhancer of zeste homolog 2 (EZH2) is crucially involved in epigenetic silencing by acting as a histone methyltransferase. Although EZH2 is overexpressed in many solid cancers, the role of EZH2 in B-cell acute lymphoblastic leukemia (B-ALL) remains largely unexplored. In a microarray experiment, we found that EZH2 was significantly upregulated in Nalm-6 cells and this was associated with the silencing of tumor suppressor genes p21, p53 and phosphatase and tensin homolog (PTEN). The abnormal expression of these genes was further confirmed by quantitative realtime polymerase chain reaction and Western blot analysis on Nalm-6 cells. Chromatin immunoprecipitation assay showed that EZH2 and H3K27me3 were both enriched in the promoter region of PTEN and p21 in Nalm-6 cells but not in normal B cells. Functional analysis showed that siRNA-mediated EZH2 knockdown led to decreased proliferation and increased apoptosis of Nalm-6 cells, accompanied by the reactivation of PTEN and p21 expression. Furthermore, we found that EZH2 inhibitor deazaneplanocin A promoted vincristine sulfate-induced apoptosis of Nalm-6 cells. Taken together, our data suggest that EZH2 is overexpressed in B-ALL and promotes the progression of B-ALL by directly mediating the inactivation of tumor suppressor genes p21 and PTEN, and could serve as a potential epigenetic target for B-ALL therapy.
Introduction
Epigenetic modifications have emerged as key mechanisms in the regulation of gene expression that are independent of changes in the primary DNA sequence, including DNA methylation, covalent histone modifications and microRNAs. Unlike DNA methylation that can provide a stable gene silencing mechanism, histone modifications can be either activation or repression markers depending on the modified residues and the type of modifications. Acetylation of the histones H3 and H4 is always connected to general transcriptional activation, and histone methylation is connected to transcription activation or repression depending on the marked positions. While the methylations on H3K4, H3K36 and H3K79 are associated with transcriptional activation, the methylations on H3K9, H3K27 and H4K20 are frequently responsible for the repression of gene transcription. 1
Histone methyltransferase enhancer of zeste homolog 2 (EZH2) is a member of the polycomb group proteins, and together with embryonic ectoderm development protein and suppressor of Zeste (SUZ12), forms the polycomb repressor complex 2, which plays an important role in a variety of biological processes, including differentiation, maintaining cell identity and proliferation, and stem-cell plasticity. 2 EZH2 contains the histone methyltransferase activity and mediates transcriptional repression of target genes at the chromatin level by catalyzing trimethylation of histone H3 lysine 27 (H3K27me3). 3 It has been shown that EZH2 is overexpressed in various types of solid cancers and hematologic malignancies. 4 High expression of EZH2 has been linked to aggressive tumor formation and poor prognosis of gastric cancer. 5 In terms of mechanism, EZH2 may contribute to the metastasis of gastric cancer through the downregulation of E-cadherin by mediating histone H3 methylation. 6 In addition, efficient inhibition of EZH2 expression results in significantly decreased cell proliferation and increased G2-M arrest. 7
B-cell acute lymphoblastic leukemia (B-ALL) is the most common malignancy in children, accounting for 30% of all cancers and 80% of all leukemias, and is caused by recurrent genetic abnormalities, including chromosomal translocations, aneuploidies and gene-specific alterations that block precursor B-cell differentiation and drive aberrant cell proliferation and survival. Chemotherapy is currently the primary treatment for this fatal disease. The effect of conventional chemotherapy regimens is mainly limited by drug toxicities. The reversible characteristics of epigenetic abnormalities have made epigenetic therapy a promising approach for B-ALL treatment. Recently, the global histone methylation inhibitor deazaneplanocin A (DZNep) was reported to selectively inhibit the expression levels of polycomb proteins and reduced trimethylation of lysine 27 on histone H3 (H3K27me3). Interestingly, DZNep treatment could lead to the reactivation of epigenetically silenced genes in breast cancer cells. 8 These results suggest that EZH2 antagonists would be promising candidates for epigenetic therapy.
However, the role of EZH2 in B-ALL remains largely unexplored, hindering the development of EZH2-based epigenetic therapy for this disease. In a microarray experiment to compare gene expression profiling between the B-ALL Nalm-6 cell line and the normal B-cell line, we found that EZH2 was significantly upregulated in Nalm-6 cells. Functional analysis showed that EZH2 knockdown led to decreased proliferation and increased apoptosis of Nalm-6 cells and this was associated with the reactivation of phosphatase and tensin homolog (PTEN) and p21 expression. Furthermore, we found that EZH2 inhibitor DZNep promoted the chemosensitivity of Nalm-6 cells, suggesting that EZH2 is a potential target for epigenetic therapy of B-ALL.
Materials and methods
Cell culture and drug treatment
The normal B-cell line used in this study was the Epstein–Barr virus transformed B lymphocyte line derived from healthy donors. The normal B cells and acute lymphoblastic leukemia cell line Nalm-6 were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin in a humidified atmosphere containing 5% CO2 at 37°C. For drug treatment, cells were passaged on the day before the drug treatment. Logarithmically growing normal B cells and Nalm-6 cells were treated with 1 μmol/L DZNep and different concentrations of vincristine sulfate (VCN; Sigma, St Louis, MO, USA) for 72 h and the cells were harvested for the following analyses.
Microarray analysis
For gene expression analysis, total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and further purified with RNeasy columns (Qiagen, Hilden, Germany) following the manufacturers’ protocols. Agilent oligo 4 × 44K human expression arrays (Agilent Technologies Inc., Palo Alto, CA, USA) were hybridized, washed and scanned according to the manufacturer's protocol. Probes/genes were considered differentially expressed if the average P value from multiple hybridizations was less than 10−6. Heat maps were generated by hierarchical clustering using GeneSpring GX software (Agilent Technologies Inc.) with color saturation as indicated.
Quantitative realtime polymerase chain reaction
Total RNA was extracted from the cells using TRIzol reagent (Invitrogen) and treated with DNase I following the manufacturer's protocol. Reverse transcription reactions were done on 1 mg total RNA by using Oligo(dT) and reverse transcriptase (Qiagen) according to the manufacturer's protocol. Quantitative realtime polymerase chain reaction (PCR) assay was performed using an iCycler thermal cycler (Bio-Rad, Hercules, CA, USA). The primers were as follows: EZH2, 5′ TGCAGTTGCTTCAGTACCCATAAT 3′ and 5′ ATCCCCGTGTACTTTCCCATCATAAT 3′; PTEN, 5′ AAGACAAAGCCAACCGATAC 3′ and 5′ GAAGTTGAACTGCTAGCCTC 3′; SUZ12, 5′ GATAAAAACAGGCGCTTACAGCTT 3′ and 5′ AGGTCCCTGAGAAAATGTTTCGA 3′; p53, 5′ CACTGCCCAACAACACCAGCTCCT 3′ and 5′ GTCTGAGTCAGGCCCTTCTGTCTTG 3′; p21, 5′ CGCTAATGGCGGGCTG and CGGTGACAAAGTCGAAGTTCC 3′; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 5′ GGTCACCAGGGCTGCTTTTA and GAGGGATCTCGCTCCTGGA 3′. SYBR Green (Takara, Dalian, China) was used as a double-stranded DNA-specific fluorescent dye. GAPDH was used as a housekeeping gene for standardization. Data were analyzed by using the 2-ΔΔCt method.
Western blot analysis
Cells were lysed in 1 mL ice-cold lysis buffer (0.1 mol/L Tris-HCl pH 8.0, 0.15 mol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid, 1% NP-40; 1 × protease inhibitor mixture, 10 mmol/L NaVO3, 1 mol/L NaF, 100 mmol/L phenylmethylsulfonyl fluoride) for 20 min. Cell lysates were further sonicated for 20 s using a Branson sonicator (Branson Ultrasonic Corporation, Danbury, CT, USA) to release histones from chromatin and the protein concentration was measured with the bicinchoninic acid assay (Biyuntian, Shanghai, China). Equal amounts of protein were separated by 10–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Whatman, Dassel, Germany). The membranes were blocked with 3% milk in 1 × TBST (0.05% Tween 20/100 mmol/L NaCl/10 mmol/L Tris-HCl, pH 7.8) blocking solution, and probed with the corresponding primary and secondary antibodies. The primary antibodies used were as follows: p21 (C-20; Santa Cruz Biotechnology, Santa Cruz, CA, USA), p53 (OP43; Oncogene Research Products, San Diego, CA, USA), μ-actin (Sigma), PTEN (BD559600; BD Biosciences, San Jose, CA, USA) and EZH2 (CST-3147; Cell Signal Technology, Danvers, MA, USA). Finally, the membranes were developed with enhanced chemiluminescence plus Western Blotting Detection System (Pierce, Rockford, IL, USA).
RNA interference
Transfections were performed by nuclear transfection using the Nucleofector system (Amaxa Biosystems, Cologne, Germany) according to the manufacturer's instructions. siRNAs targeting Ezh2 5′-AAGACTCTGAATGCAGTTGCT-3′ and mock siRNA 5′-CTTACGCTGAGTACTTCGA-3′ were synthesized by GenePharma (Shanghai, China). At 72 h after transfection, the cells were harvested and subjected to Western blot and reverse transcription (RT)-PCR analysis.
Flow cytometry analysis of apoptosis
Apoptosis was measured by flow cytometry using propidium iodide (PI)/annexin V-fluorescein isothiocyanate staining (BD Biosciences). In brief, cells were washed with ice-cold phosphate buffer saline and resuspended in 200 μL binding buffer. The cells were mixed with 10 μL annexin V stock solution and incubated for 30 min. Next, the cells were stained with 5 μL PI and finally analyzed on a BD FACScalibur equipped with CellQuest software (BD Biosciences).
Cell growth assay
After nuclear transfection, the cells were stained with trypan blue. A cell growth curve was drawn by plotting cell number against culture time. Cell numbers were counted in triplicate at defined times.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) experiments using antibodies against EZH2 (CST3147) and H3K27me3 (Cat No. 07-449; Upstate-Millipore, Billerica, MA, USA) were performed using the EZ ChIP™ kit (Millipore) according to the manufacturer's protocol. Nalm-6 and normal B cells were crosslinked in 1% formaldehyde for 10 min at room temperature and were then lysed and sonicated with the Branson 250D Sonifier to obtain chromatin fragments between 200 and 700 bp. After dilution and preclearing, whole-cell lysates were incubated with antibodies overnight at 4°C. Protein A agarose beads (Upstate-Millipore) were used to pull down antibody-chromatin complexes, which were then washed with low-salt, high-salt, LiCl and TE (pH 8.0) wash buffers. Enriched chromatin fragments were eluted, subjected to crosslink reversal at 65°C for 6 h and purified with a Qiagen DNA extraction kit. The primers for ChIP PCR were as follows: p21, GGTGTCTAGGTGCTCCAGGT and GCACTCTCCAGGAGGACACA; PTEN, AAACGAGCCGAGTTACCG and GACTGCATTCGCTCTTTCCT.
Statistical analysis
Results are presented as mean ± standard deviations for indicated experiments. Statistical significance was estimated by Student's t-test with SPSS 11.5 software (SPSS Inc., Chicago, IL, USA). P values of less than 0.05 were considered to indicate statistical significance.
Results
Comparative analysis of gene expression profiling between Nalm-6 and normal B cells
First, we performed a microarray experiment to compare gene expression profiling between Nalm-6 and normal B cells. The hierarchical clustering analysis of the transcriptomes of Nalm-6 and normal B cells showed that several tumor suppressor genes such as p21, p53 and PTEN were silenced, while genes involved in the epigenetic regulation, such as histone deacetylases, DNA methyltransferases and polycomb proteins, were significantly upregulated in Nalm-6 cells (Supplementary Figure 1, please see The differentially expressed genes between Nalm-6 and normal B cells. (a, b) Quantitative realtime polymerase chain reaction analysis of enhancer of zeste homolog 2 (EZH2), SUZ12 (a), p21, p53 and phosphatase and tensin homolog (PTEN) (b) mRNA levels in Nalm-6 and normal B cells (n = 3). (c) Western blot analysis of EZH2, p21, p53 and PTEN protein concentrations in Nalm-6 and normal B cells. Actin served as loading control. Shown are representative blots from three independent experiments with similar results
EZH2 knockdown inhibits the growth and induces the apoptosis of Nalm-6 cells
Given the upregulation of EZH2 expression in Nalm-6 cells, we speculated on the biological consequences of EZH2 upregulation. We therefore employed a loss of function approach to knockdown EZH2 in Nalm-6 cells. After Nalm-6 cells were transfected with specific siRNA against EZH2 and mock siRNA as a control, we observed that a reduced EZH2 protein level was accompanied by a decreased histone H3K27me3 level in EZH2 siRNA, but not in mock siRNA-transfected cells (Figure 2a), confirming the effective knockdown of EZH2. Next, we examined the effect of EZH2 knockdown on the growth of Nalm-6 cells and found significantly inhibited growth of Nalm-6 cells transfected with EZH2 siRNA compared with mock siRNA-transfected cells (Figure 2b). In addition, annexin V staining and fluorescence-activated cell sorting analysis demonstrated that EZH2 knockdown led to a significant increase of apoptotic cells (P < 0.05, n = 3; Figures 2c and d). These results suggest that EZH2 upregulation contributes to Nalm-6 cell growth via the inhibition of apoptosis.
Enhancer of zeste homolog 2 (EZH2) knockdown inhibits the growth and induces the apoptosis of Nalm-6 cells. (a) EZH2 protein and H3K27me3 concentrations were obviously decreased in cells transfected with EZH2 siRNA compared with untransfected or mock siRNA-transfected cells. Shown are representative blots from three independent experiments with similar results. (b) The growth curve of Nalm-6 cells transfected with mock or EZH2 siRNA. (c) Representative flow histograms of fluorescence-activated cell sorting analysis of apoptotic Nalm-6 cells after transfection with mock or EZH2 siRNA. (d) Percentage of apoptotic Nalm-6 cells after transfection with mock or EZH2 siRNA (n = 3). *P < 0.05 versus mock transfected cells. PI, propidium iodide. (A color version of this figure is available in the online journal)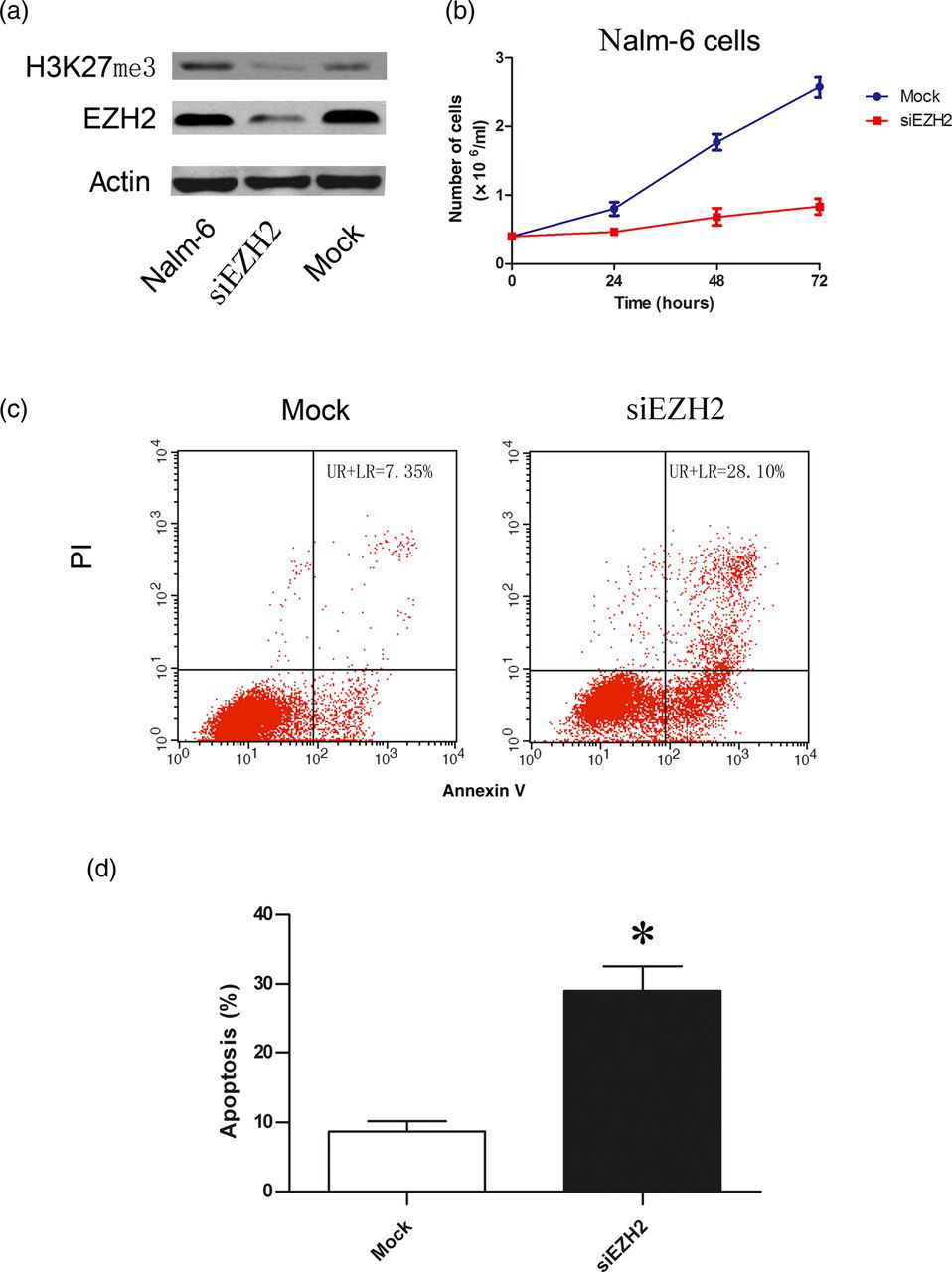
EZH2 knockdown reactivates PTEN and p21 expression in Nalm-6 cells
To investigate whether EZH2 knockdown could reactivate the expression of tumor suppressor genes, we performed quantitative RT-PCR and Western blot analysis. The results showed that the expressions of PTEN and p21 at both mRNA and protein concentrations were significantly higher in Nalm-6 cells transfected with EZH2 siRNA than in the cells transfected with mock control siRNA (Figures 3a and b), but p53 expression was not significantly increased after EZH2 knockdown (data not shown).
Enhancer of zeste homolog 2 (EZH2) knockdown reactivates the expression of p21 and phosphatase and tensin homolog (PTEN) in Nalm-6 cells. (a) Quantitative realtime polymerase chain reaction (PCR) analysis of p21, p53 and PTEN mRNA levels in Nalm-6 transfected with mock or EZH2 siRNA (n = 3). *P < 0.05 versus mock transfected cells. (b) Western blot analysis of p21 and PTEN protein concentrations in Nalm-6 transfected with mock or EZH2 siRNA. Actin served as loading control. Shown are representative blots from three independent experiments with similar results. (c) Chromatin immunoprecipitation (ChIP)-PCR analysis showing the binding of EZH2 and H3K27me3 to the promoter region of PTEN in Nalm-6 cells but not in normal B cells. (d) ChIP-PCR analysis showing the binding of EZH2 and H3K27me3 to the promoter region of p21 in Nalm-6 cells but not in normal B cells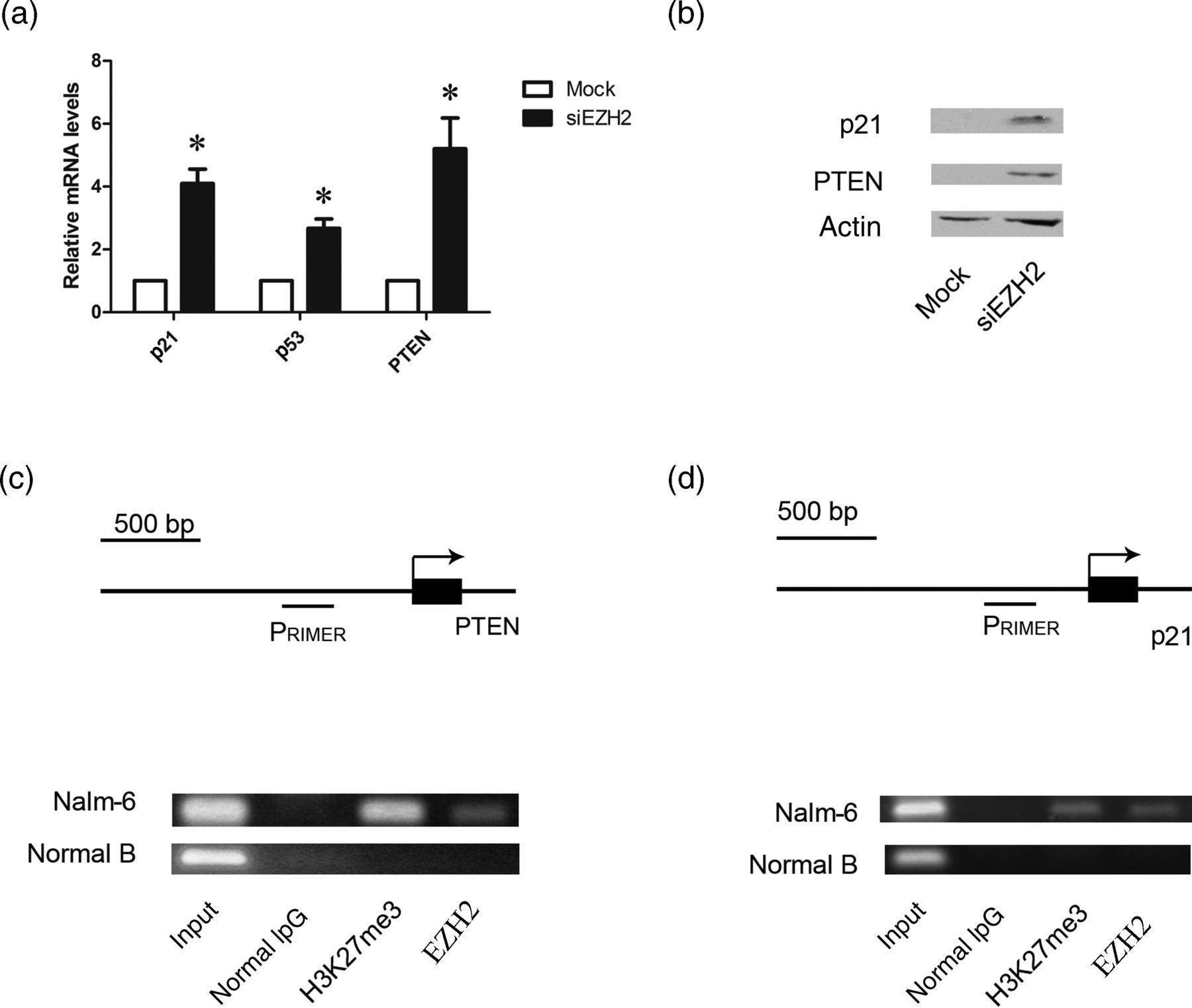
These results prompted us to investigate if the silencing of PTEN and p21 is directly regulated by EZH2 in Nalm-6 cells. Therefore, we performed a ChIP assay in normal B cells and Nalm-6 cells using histone H3K27 trimethylation antibody and EZH2 antibody. Equal amounts of non-specific normal rabbit IgG were used as controls for the specificity of the ChIP assay. The results showed that EZH2 and H3K27me3 were both enriched in the promoter region of PTEN and p21 genes in Nalm-6 cells but not in normal B cells (Figures 3c and d). These results suggest that polycomb protein EHZ2 promotes the epigenetic inactivation of PTEN and p21 expression in Nalm-6 cells.
EZH2 inhibitor DZNep promotes the chemosensitivity of Nalm-6 cells
To further explore the clinical significance of our findings, we treated Nalm-6 cells with the specific EZH2 inhibitor DZNep and examined their chemosensitivity. First, we confirmed that the dose of 1 μmol/L DZNep was effective to reduce Ezh2 expression in Nalm-6 cells (Figures 4a and b). Next, we treated Nalm-6 cells with DZNep and VCR and found that DZNep treatment led to a two–three-fold increase in VCR-induced apoptosis of Nalm-6 cells (Figure 4c). These data suggest that EZH2 functions to protect Nalm-6 cells against chemotherapy-induced apoptosis.
Deazaneplanocin A (DZNep) promotes vincristine sulfate (VCR)-induced apoptosis of Nalm-6 cells. (a) Nalm-6 cells were treated with 100 ng/mL VCR in combination with 1 μmol/L DZNep or dimethyl sulfoxide (DMSO) for 72 h, and the expression of enhancer of zeste homolog 2 (EZH2) was detected by Western blotting. Actin served as loading control. (b) Percentage of apoptotic Nalm-6 cells determined by flow cytometry after treatment with VCR as indicated in combination with 1 μmol/L DZNep or DMSO (n = 3). *P < 0.05 versus DMSO-treated cells at the indicated concentration of VCR. (c) Quantization of EZH2 protein expression based on (a). *P < 0.05 versus untreated cells or DMSO-treated cells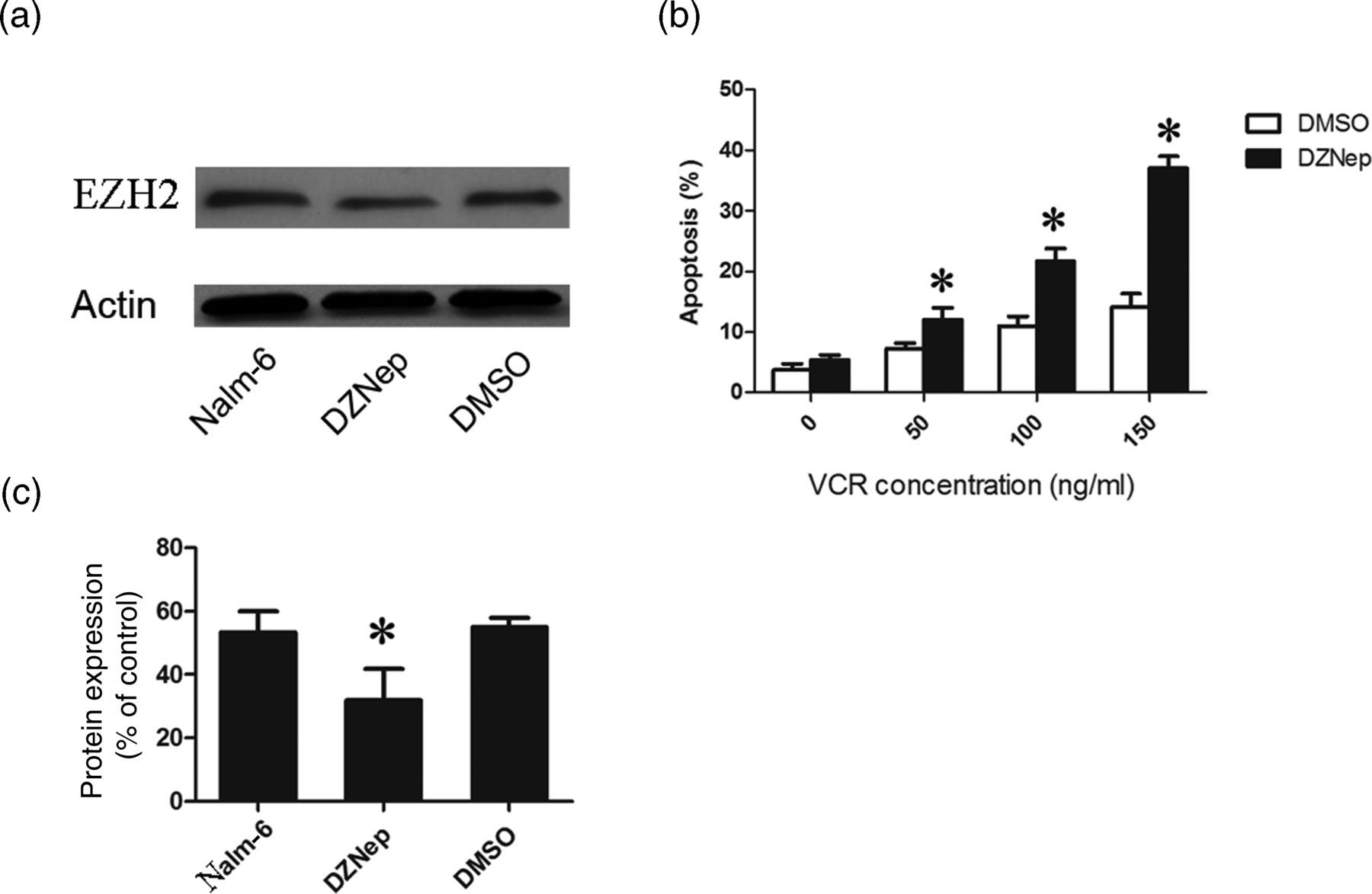
Discussion
In the past few years, an increasing body of evidence has proven that the inactivation of tumor suppressor genes by epigenetic alterations, including DNA hypermethylation and histone hypoacetylation, is an important mechanism involved in carcinogenesis. 9 In this study, we mainly focused on EZH2, the major enzyme that methylates histone H3 lysine 27 (H3K27). EZH2 is a member of the polycomb group of proteins, and plays a crucial role in cancer initiation and progression. 4 High expression of EZH2 has been shown to be associated with upregulated cell proliferation and invasion, cancer metastasis and poor prognosis in various malignancies. 4
When we performed a comparative microarray analysis of gene expression profiling between B-ALL Nalm-6 cells and normal B cells, we found that the expression of EZH2 was higher in Nalm-6 cells than in normal B cells, and this was associated with the downregulation of some tumor suppressor genes such as p21, p53 and PTEN in Nalm 6 cells. These results were further confirmed by quantitative realtime PCR and Western blot analysis. Therefore, we conclude that EZH2 is overexpressed in B-ALL cells.
Previous studies have demonstrated that several tumor suppressor genes such as DAB2IP, PSP94, p16 INK4A/ARF, p57 and Bim are directly repressed by EZH2 through histone H3K27 trimethylation around the promoter regions and could be reactivated upon the depletion of EZH2. 10–13 Thus, we wondered if there is a direct link between the overexpression of EZH2 and the silencing of tumor suppressor genes p21, p53 and PTEN in B-ALL Nalm-6 cells. The ChIP experiment demonstrated that the promoter regions of PTEN and p21 were enriched with H3K27me3 and EZH2 in Nalm-6 cells but not normal B cells. Next, we observed that siRNA-mediated knockdown of EZH2 could restore the expressions of p21 and PTEN at both mRNA and protein levels in Nalm-6 cells. These results provide strong evidence that an epigenetic mechanism mediated by Ezh2 contributes to the silencing of tumor suppressors p21 and PTEN in B-ALL. However, further studies are necessary in the future to characterize the mechanism by which EZH2 is upregulated in B-ALL.
p21 (CDKN1A) is a cyclin-dependent kinase inhibitor that plays a pivotal role in cell cycle arrest induced by HDAC inhibitor treatment in many cancer cell lines. 14 PTEN is a tumor suppressor with lipid and protein phosphatase activity that negatively regulates receptor tyrosine kinase signaling. PTEN is essential for the prevention of leukemogenesis, and conditionally deleting PTEN in adult hematopoietic cells led to myeloproliferative disease within days and transplantable leukemia within weeks. 15 Moreover, it was reported recently that PTEN overexpression could delay the development of B-ALL in mice. 16 Consistent with the reactivation of p21 and PTEN expression after EZH2 knockdown in Nalm-6 cells, we observed that cell growth was inhibited and apoptosis was promoted. Furthermore, inhibition of EZH2 by DZNep promoted VCR-induced apoptosis of Nalm-6 cells. Interestingly, a recent study showed that the inhibition of EZH2 exhibited an anticancer effect for non-small cell lung cancer cells due to the upregulation of the expression of tumor suppressor E-cadherin. 17 These data suggest that due to the silencing of tumor suppressor genes, EZH2 increases the cell resistance to chemotherapy and promotes the development of B-ALL. However, EZH2 is known to be inactivated in myeloid malignancy due to gene mutation. 18 Therefore, the different function of EZH2 in myeloid or lymphoid malignancies may partially depend on the status of p53.
Epigenetic abnormalities play pivotal roles in tumorigenesis. Due to the reversible nature of epigenetic modification, it has been suggested that epigenetic drugs could be a new direction in cancer therapy. 19 Our results on the synergistic effects of EZH2 inhibitor and VCR to promote the apoptosis of B-ALL cells support this proposal. It is known that cancer cells develop resistance to chemotherapy through epigenetic silencing of tumor suppressor genes and proapoptosis factors. 20 Combination of epigenetic therapy with standard chemotherapy could prevent the development of chemoresistance and improve the cure rates of malignancies including B-ALL.
Footnotes
ACKNOWLEDGMENTS
This study was supported by grants from Natural Science Foundation of China (Nos 30830052, 30911130363), National Key Scientific Program of China (No. 2011CB964901), Program for Changjiang Scholars and Innovative Research Team in University-PCSIRT (No. IRT0909).