
Abstract
The potential link between the inflammatory effects of postprandial lipemia and the induction of macrophage foam cell formation by triacylglycerol-rich lipoproteins (TGRL) was studied using postprandial triacylglycerol-rich lipoproteins (ppTGRL) derived from human volunteers and primary human monocyte-derived macrophages (HMDM). Subjects were fed a test meal high in dairy fat, followed three hours later by isolation of serum ppTGRL. Pro-inflammatory (M1) and anti-inflammatory (M2) phenotypes were induced in HMDM by treatment with lipopolysaccharide (LPS) or dexamethasone (DEX), respectively. ppTGRL caused a dose-dependent increase in both triacylglycerol (TG) and cholesterol (CH) accumulation in the cells. TG accumulation was unaffected by LPS or DEX treatment, but LPS as compared with DEX-treated HMDM were found to accumulate more CH, and this effect was greater than that induced by ppTGRL in untreated cells. LPS-treatment had no effect on lipid uptake from ppTGRL (via the LDLr, scavenger receptors or SR-B1) or on CH efflux, but the CH synthesis inhibitor mevinolin abolished the difference between CH accumulation in LPS-and DEX-treated cells, suggesting that CH synthesis is enhanced in the inflammatory state. Phospholipid (PL) synthesis was increased in inflammatory M1 as compared with anti-inflammatory M2 HMDM. Moreover, TG synthesis was decreased by ppTGRL in DEX-treated as compared with untreated cells. We conclude, therefore, inflammation causes a greater increase in the accumulation of neutral lipids than ppTGRL in macrophages, and that this effect is related to modulation of PL metabolism and possibly also CH synthesis. Thus, the inflammatory phenotype of macrophages influences their lipid metabolism, and is, therefore, likely to modulate the induction of macrophage lipid accumulation by lipoproteins associated with foam cell formation.
Keywords
Introduction
It is now well established that inflammation is a potent contributor to the development of atherosclerotic lesions, but the direct relationship between the two processes is largely unexplored. Inflammation and atherosclerosis are initiated when macrophages which have migrated into the artery wall, take up lipid from the plasma lipoproteins and become foam cells.1–3 The lipoproteins involved in atherogenic processes include both LDL and chylomicron remnants. There is general consensus that the internalization of LDL, and particularly of oxidized-LDL, mediated by scavenger receptors, plays a role in foam cell formation and atherogenesis.4,5 On the other hand, however, there is increasing evidence that lipoproteins carrying dietary lipids are also pro-atherogenic. 6
Postprandial (pp) lipemia is the transient increase in blood lipids which occurs after a meal containing fat, and is caused by raised levels of triacylglycerol-rich lipoproteins (TGRL), including chylomicrons, chylomicron remnants and very-low-density lipoprotein (VLDL), in the blood. Lipids from the diet are absorbed and packaged in the intestine into large lipoproteins, rich in triacylglycerol (TG), called chylomicrons. These pass into the blood and are rapidly lipolysed by lipoprotein lipase (LPL), forming the smaller chylomicron remnants, which deliver the remaining TG and the other dietary lipids to the liver. 7 Current evidence suggests that ppTGRL may be involved in promoting atherosclerosis both directly, via an influence on events in the vasculature, and indirectly, by causing a more atherogenic lipoprotein profile when their levels are abnormally raised. Delayed clearance of dietary lipids leads to accumulation of circulating ppTGRL, and there is now a great deal of evidence to support the idea that this condition is associated with premature atherosclerosis.6,8,9 Furthermore, lipo-proteins of intestinal origin have been isolated from atherosclerotic plaque 10 and it has been showed that TGRL cause endothelial dysfunction and macrophage foam cell formation.6,11,12
In addition to their role as precursors of foam cells, macrophages are immune, infection and inflammatory1–3,13 mediator cells secreting a large array of chemotactic molecules, lipases and cytokines which influence the entire process of atherosclerosis development consistently, due to their presence in the lesions during all stages of athero-genesis.2,4,5 The phenotype and function of arterial wall-resident macrophages are regulated by various inflammatory and immune stimuli, which may induce macrophage activation (M1 phenotype) or, alternatively, an anti-inflammatory or M2 phenotype. 14 Pro-activated M1 macro-phages are believed to display increased function/activity of lipoprotein receptors leading to an increased induction of lipid accumulation.15–18 However, almost nothing is known about the role of the M1 or M2 macrophage pheno-type in their response to ppTGRL. The aim of this study was to investigate the effects of the inflammatory phenotype of macrophages on their lipid metabolism and to determine how this is affected by ppTGRL. We also investigated the mechanisms which contribute to the observed effects of ppTGRL on macrophages. Lipopolysaccharide (LPS) and dexamethasone (DEX), respectively, were used to induce pro-inflammatory (M1) and anti-inflammatory (M2) pheno-types in primary human macrophages and ppTGRL were obtained from normo-lipidemic volunteers after a test meal.
Materials and methods
[1(3)-3H]glycerol (30–60 mCi/mmol), [1α, 2α(n)-3H]chol-esterol oleate (30–60 mCi/mmol), [4-14C]cholesteryl-oleate (60 mCi/mmol), [1α, 2α(n)-3H] cholesteryl oleyl ether (30–60 mCi/mmol), cholesteryl[1-14C]oleate (30–60 mCi/ mmol) and [14C]phosphatidylcholine were supplied by GE Healthcare, Bio-Sciences (Milan, Italy). Iscove's Dulbecco's modified medium (IMDM), fetal bovine serum (FBS), and Ficoll-Paque were obtained from Hyclone Europe Ltd (Sial, Rome, Italy). FBS, Ficoll-Paque, penicillin and streptomycin were supplied from EuroClone Europe Ltd (Sial, Rome, Italy). CD14 MicroBeads and LS Separation Columns were purchased from Miltenyi Biotec (Bologna, Italy). Monoclonal antibodies phycoerythrin (PE) or allo-phycocyanin (APC)-Cy7 conjugated anti-CD14, APC-conjugated anti-CD68, PE-conjugated anti-CD206 and IgG isotypes were purchased from Becton Dickinson, Bio-sciences (Buccinasco, Italy), and flow cytometry staining buffer, fixation buffer and permeabilizing solution were obtained from ebioscience (Prodotti Gianni, Milan). Full-length LPS from Escherichia coli serotype 0111:B4, fatty acid-free bovine low endotoxin serum albumin (BSA), DEX, mevinolin, orlistat and various classes of lipids and commercial solvents and reagents were purchased from Sigma-Aldrich, Milan, Italy. WAY-121898 (way) was obtained from Wyeth Pharmaceutical and Research (Philadelphia, PA, USA). BLTs were obtained from Chem-Bridge Corporation (San Diego, CA, USA). Tumor necrosis factor-α (TNF-α) and interleukin-10 (IL-10) enzyme-linked immnunosorbent assay (ELISA) kits were obtained from Bender System (Milan, Italy). The study was approved by the Ethical Committee of the Istituto Superiore di Sanità.
Flow cytometry
Cell surface and intracellular phenotyping of monocytes and human monocyte-derived macrophages (HMDM) was performed using direct immunofluorescence assays as described before with minor modifications. 19 To evaluate CD14 expression, in monocytes and HMDM, cells were washed with IMDM + FBS (15% v/v) (IMDM + FBS), incubated in fetal bovine serum for 20 min on ice for Fc receptor (FCR) blocking and then stained for 30 min at 4°C with the monoclonal antibody PE-conjugated anti-CD14 or IgG iso-types, diluted 1:10 in IMDM + FBS. After two washings with cold IMDM + FBS, cells were fixed in paraformaldehyde 2% and analyzed using a fluorescence-activated cell sorter (FACS) (Becton Dickinson).
After treatment of HMDM with LPS and DEX as described below, cells were washed and blocked for FCR as described above. For cell surface staining, HMDM were washed with staining buffer and then incubated for 30 min with APC-Cy7 conjugated anti-CD14 or PE-conjugated anti-CD206. For staining of CD68, which is predominantly an intracellular protein (although smaller amounts can be detected on the cell surface), after cell surface staining, HMDM were fixed with fixation buffer for 20 min at room temperature, permeabilized with FACS permeabilizing solution for 10 min, then washed and stained by incubation for 20 min at 4°C with APC-conjugated anti-CD68. Appropriate isotype-matched antibodies were used as negative controls for non-specific binding (negative controls) of cell-surface and intracellular staining. Immunofluorescence was assessed by flow cytometry (Becton Dickinson).
HMDM and manipulation of their inflammatory phenotype
Buffy coats from the blood of healthy donors were diluted 1:3 with phosphate-buffered saline (PBS) and layered on Ficoll-Paque. After centrifugation, white blood cells were collected and washed with PBS. CD14 MicroBeads were used to positively select human monocytes from the white blood cells according to the manufacturer's instructions. Monocytes (CD14-positive fraction) were washed, transferred to 22-mm dishes (1.5 × 10 6 cells; 8 × 105 cells/mL) and cultured in IMDM + FBS. The purity of isolated monocytes, monitored by specific flow cytometric analysis for CD14, was 95–97%. Experiments were performed with HMDM 10 d after plating. 21
Macrophage pro-inflammatory (M1) and anti-inflammatory (M2) phenotypes were induced by treatment with LPS and DEX, respectively. Ten days after isolation, HMDM were incubated for 18 h in complete IMDM alone (untreated, NT) or complete IMDM containing LPS (1 μg/mL) or DEX (0.1 μmol/L). After these pretreatments, HMDM were incubated in the presence of ppTGRL and/or other factors at under the conditions indicated.
Lipoproteins
ppTGRL were isolated from normo-lipidemic volunteers three hours after consumption of fatty breakfast meal, according to Jackson et al., 22 with some modifications. The aim of the study was clearly explained to the subjects and formal consent was obtained from each. Subjects were informed of the results obtained with their specimen. For most subjects more than one blood sample was obtained during the three-year study. A total of nine subjects (5 female and 4 male) aged 36.6 ± 3.2 years participated. The subjects were of the same ethnic group, with similar dietary regimen (Mediterranean diet) and life-style, did not use any dietary supplements and were not under therapy, and acute or chronic recognized diseases were absent during the study. The breakfast meal consisted of 200 g whole milk, six sweet biscuits, 60 g mixed fruit jam, 50 g butter, 100 g cream and coffee and was consumed within 20 min. The total energy intake was 1050 kcal containing 65%, 29% and 6% fat, carbohydrate and protein, respectively. Blood samples were collected into heparinized tubes, centrifuged at 2500 rpm for 15 min at 4°C to obtain the plasma, and penicillin (100 U/mL), streptomycin (100 μg/mL) and phenylmethanesulfonyl fluoride 60 μg/mL were added. 23 Four milliliters of plasma, adjusted to a density of 1.1 g/mL with KBr, was placed at the bottom of an albumin-coated (5% w/v in saline) centrifuge tube, and over layered with KBr solutions of density: 1.063 g/mL (1.75 mL), 1.019 g/mL (1.75 mL) and 1.006 g/mL (3 mL). Ultracentrifugation was performed at 30,000 rpm in a SW 40 Ti swinging-bucket rotor for three hours at 15°C. Lipid particles (ppTGRL, d < 1.006 g/mL) were harvested from the top layer. All ppTGRL were used the day after preparation.
For some types of experiment ppTGRL obtained from a single donor was not sufficient to perform all the analyses planned. In addition, ppTGRL cannot be easily labeled in their lipid components to specifically follow their individual fate. For this reason, some experiments were performed with an experimental model of pp lipoprotein such as chylomicron remnant-like particles (CRLP). CRLP, [3H]cholesteryl oleate ([3H]CE)- and [3H]cholesteryl ether ([3H]Cether)-labelled CRLP were prepared as previously described.21
After separation, ppTGRL and CRLP were dialyzed overnight against IMDM containing penicillin/streptomycin, kept in the dark at 4°C under nitrogen and used within two days. TG, unesterified (UC) and total cholesterol (CH) content of the lipoproteins were determined by routine methods.
[3H]glycerol test
Synthesis of TG and phospholipid (PL) in macrophages was evaluated using [3H]glycerol as previously described.24 NT, LPS- or DEX-treated HMDM were incubated for six hours at 37°C in serum-free IMDM containing ppTGRL (0,10, 30 μg CH/mL) in the presence of [3H]glycerol (4 μCi/mL, 20 μmol/L). After the incubations, HMDM were washed three times with PBS and lipids were extracted by hexane/isopropanol; 3:2, v/v. [14C]CE was added as an internal standard and the lipids classes were separated by thin-layer chromatography on silica gel (Merck, Darmstadt, Germany) developed in hexane/ether/acetic acid (70:30:1, v/v/v). Radioactivity associated with the bands corresponding to [3H]TG and [3H]PL was assayed in an LS5000 Beckman liquid scintillation counter.
LDL from plasma from fasted normo-lipidemic volunteers was radiolabelled with [3H]CE to prepare [3H]CE-LDL (1.019-1.063 g/mL) (specific activity; 670 ± 120 dpm/ nmol) or used to obtain [3H]CE-acLDL (specific activity 771 + 99 dpm/mL) as described before.21 After 24-h treatment with LPS or the vehicle only, HMDM were washed and further incubated with [3H]CE-nLDL or [3H]CE-acLDL (100 μg protein/mL) for six hours For both LDL and acLDL, about 94% of the radiolabel was associated with CE and the remaining activity with UC in the lipoprotein preparations and cell samples. Total labeled CH taken up by HMDM was calculated as the sum of the radioactivity associated with the [3H]UC and [3H]CE bands and expressed as pmol[3H]cholesterol/h/mg of cell protein.
Cell lipid determination
For the determination of intracellular lipids, after incubation, cells were washed three times with PBS and harvested by scraping into 500 μL of distilled water. Lipids were extracted as before 25 and the CH and TG content was determined in duplicate by fluorimetric methods according to Gamble et al. 26 and Mendez et al., 27 respectively. An aliquot of cellular suspension was utilized to determine protein content by Bradford's method, 28 using BSA as a standard.
Gas chromatography
The fatty acid distribution of ppTGRL was determined by gas chromatographic analysis. 29 Briefly, lipids were extracted and the methyl esters of the total lipid extract were prepared by acid-catalyzed transesterification using heptadecanoic acid (17:0) as an internal standard. The fatty acid methyl esters were extracted in hexane, evaporated under nitrogen and analyzed by gas chromatography in a Hewlett-Packard 5890 Series II gas chromatograph (Hewlett Packard, Milan, Italy) equipped with a flame ionization response detector, a capillary column OmegawaxM 320 (30 m × 0.32 mm) and a 0.25 mm film (Supelco, Inc, Milan, Italy).
Measurements of LPL activity
LPL activity was determined by measuring the release of [3H]-fatty acid from [3H]triolein. 30 Ten days after plating, HMDM (3 × 106 cells) were washed and incubated in serum-free IMDM with 10 U/mL heparin alone (NT) or containing LPS (1 μg/mL) or DEX (0.1 μmol/L). After 18 h, the medium was collected, centrifuged to remove cell debris and stored at -80°C until analysis. The substrate was prepared as an emulsion of triolein containing 0.9 mg triolein and [3H]oleic acid labeled triolein (120,000 dpm/tube) and the solvent was removed under a stream of nitrogen. The lipids were dissolved in Tris buffer (0.4 mol/L, pH 8.4) containing Triton X-100 and then sonicated for 30 min on ice. Aliquots of medium, in duplicate, were incubated for 30 min at 28°C with 120 μL of substrate in the presence of fatty acid free-BSA (20% w/v) containing heparin (1U/tube). The reaction was stopped by the addition of 1.5 mL of chloroform/methanol/ toluene (2:2.4:1 v/v/v) containing 0.29 mmol/L oleic acid as a carrier and centrifuged to separate the organic layer and the aqueous phase. The amount of [3H]oleate released into the upper phase was counted in an LS 5000 Beckman scintillation counter.
mRNA determination
Total cellular RNA was isolated using NucleoSpin RNA (Macherey-Nagel, Duren, Germany) according to manufacturer's instructions. Quantitative realtime polymerase chain reaction (q-PCR) was performed in a two-step process (Mx3000P Instrument, Applied Biosystems, Monza, Italy). RNA was reverse-transcribed to cDNA and amplified by PCR using the AffinityScript qPCR cDNA synthesis kit from Stratagene (Stratagene, Waldbronn, Germany) and the following conditions: 25°C for five minutes, followed by 42°C for 15 min, and 95°C for five minutes. cDNA was then quantified using the SYBR Green Master Mix (Stratagene). The reactions contained 100 ng of cDNA, 100 nmol/L of gene-specific primers (Eurofins MWG Operon, ‘M-Medical, Cornaredo, Italy) (Table 1) and the SYBR Green PCR Master Mix in a total volume of 25 μL. qPCR consisted of an initial hold at 95°C for 10 min, and then 40 cycles of 95°C for 30 s and 72°C for one minutes Housekeeping PCR was performed with each new cDNA to confirm similar starting concentrations for all PCR procedures. Gene expression was normalized using glyceraldehyde-3-phosphate dehydrogenase. mRNA expression was quantified as described by Pfaffl. 31
Primer sequences and product sizes for determination of lipases mRNA expression
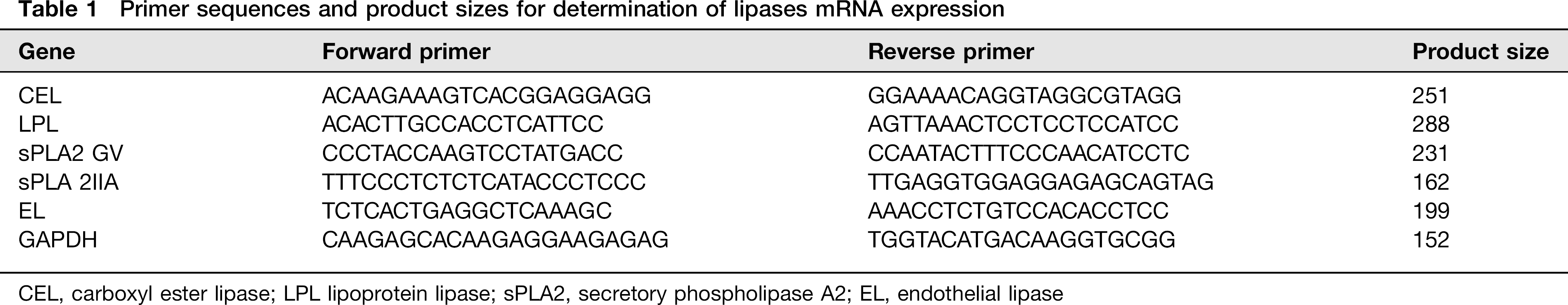
CEL, carboxyl ester lipase; LPL lipoprotein lipase; sPLA2, secretory phospholipase A2; EL, endothelial lipase
Other analytical methods
For TNF-α and IL-10 determinations, cell medium was collected, centrifuged to remove debris and stored to -80°C till analysis. ELISA assays for TNF-α and IL-10 and other cytokines were performed according to the manufacturer's instructions. Lipoprotein lipids were determined with commercial enzymatic reagent kits (Bayer and Li StarFISH, Milan, Italy). Endotoxin levels in ppTGRL preparations were measured by Limulus amoebo-cyte lysate analysis (PYROTELL, PBI, Milan, Italy).
Statistical analysis
As each experiment was performed with HMDM derived from a different individual donor, the statistical differences between experimental conditions were analyzed by repeated measures analysis of variance (RM-ANOVA). Significant differences were calculated using the Tukey– Kramer multiple comparison post hoc test or by Student's paired t-test.
Results
Characterization of HMDM after treatment with LPS or DEX
Since in some experimental conditions, macrophage expression of CD14, an essential component of the LPS receptor complex, 32 may be reduced, 33 we first tested CD14 expression in our experimental conditions using both mono-cytes and HMDM from different donors. CD14 was expressed by 93 ± 0.61% of monocytes on the day of isolation and by 52 ± 1.2% of HMDM after 10 d of culture (Figure 1). Treatment of HMDM with either LPS or DEX caused further reduction (-30% to 40%) in CD14 expression (Figure 2a). The macrophage marker CD68, which is expressed intracellularly and on the surface 20 was also highly expressed by HMDM and reduced (-25% to 40%) by both treatments (Figure 2b). In contrast, the expression of CD206, a marker of the M2 phenotype, 34 was decreased by about 50% by LPS, but increased by more than two-fold by DEX (Figure 2c).
The phenotype of monocytes, on the isolation day (day 0), and after 10 d in culture (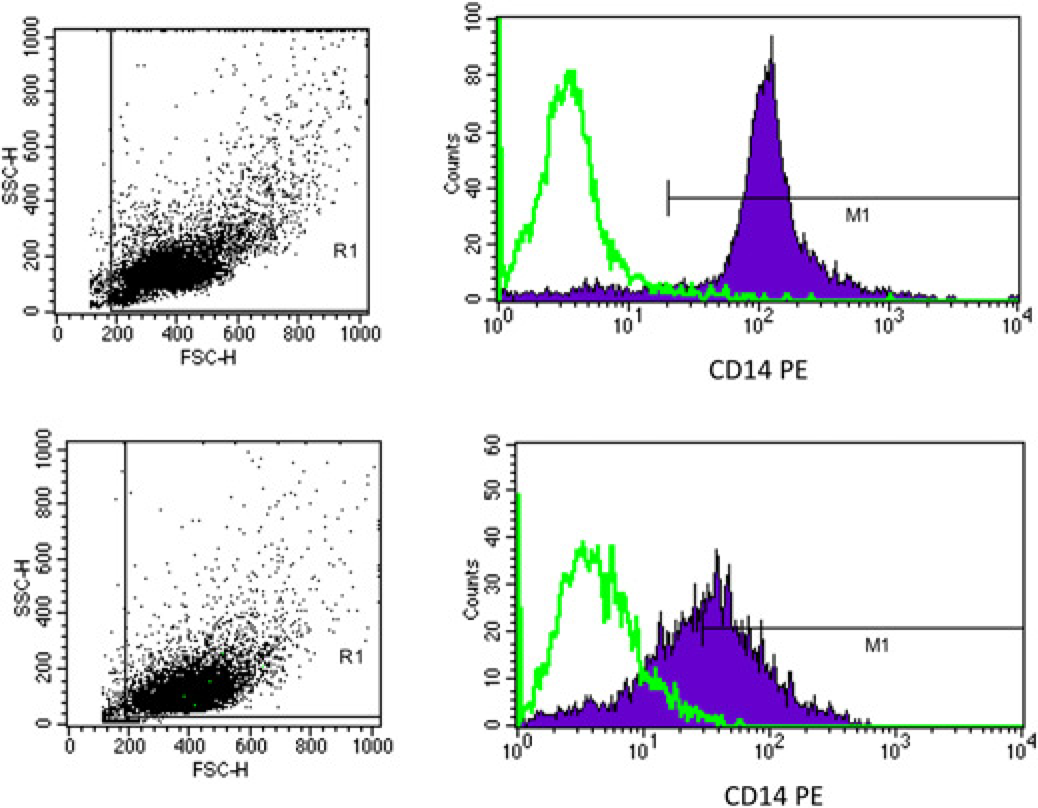
HMDM were incubated in 
We also tested the effect of LPS and DEX on HMDM by measuring the secretion of TNF-α and IL-10. The results (Figure 3) showed that, in comparison with NT cells, DEX increased IL-10 secretion by about two-fold (P < 0.05 versus NT), while the levels of TNF-α were not significantly changed. LPS, on the other hand, increased the secretion of IL-10 to a much greater extent than DEX (5-fold) (P < 0.001) and also increased TNF-α secretion by about six-fold compared with NT cells (P < 0.0001). In addition, in other experiments the production of cytokines known to be induced in M1-activated macrophages, including IL-6, IL-12, macrophage inflammatory protein (MIP)-1a, MIP-1b and monocyte chemoattractant protein (MCP)-1 35 were found to be greatly increased by exposure of HMDM to LPS (IL-6, >10-fold; IL12, 3-fold; MIP-1α, 46-fold; MIP-1β, 21-fold; MCP-1, >5-fold), but unchanged or increased to a much lesser extent after treatment with DEX (IL-6, <5-fold; IL-12, unchanged; MIP-1a, 3-fold; MIP-1b, 2.5-fold; MCP-1, unchanged). IL-8 secretion by the cells, on the other hand, was not affected by either treatment.
HMDM were incubated in 
Overall, these results indicate that LPS polarizes HMDM towards the M1 inflammatory phenotype, while DEX shifts the balance towards a more anti-inflammatory M2 phenotype.
Characterization of ppTGRL
Table 2 shows the lipid and apolipoprotein B48 content and fatty acid distribution in ppTGRL. The TG/CH molar ratio was similar to that reported for chylomicron remnants.24,36 More than 55% of total fatty acids was saturated fat, with little variation between preparations (Table 2). Endotoxin was not detectable (below 0.06 EU/mL, with US standard endotoxin) in any ppTGRL preparation used.
Lipid content, fatty acid distribution and apolipoprotein

TG, triacylglycerol; SFA, saturated fatty acid; MUFA, mono-unsaturated fatty acid; PUFA, poly-unsaturated fatty acid
Data are the mean ± SEM and the number of experiments is shown in brackets
CH from lipoproteins may enter macrophages by uptake via receptors including the LDLr or scavenger receptors, or by selective uptake of CE. 21 Uptake of [3H]CE-nLDL or [3H]CE-acLDL by NT and LPS-treated HMDM was similar ([3H]CE-nLDL; 10,300 ± 1543 (NT) and 10,652 ± 2890 (LPS): [3H]CE-acLDL; 9236 ± 2600 (NT); 13,050 ± 3100 (LPS) pmol[3H]cholesterol/h/mg protein) indicating that LPS treatment did not affect LDLr or scavenger receptor activity.
Macrophage phenotype and lipid accumulation
In order to investigated the effects of macrophage M1 and M2 phenotypes on lipid accumulation induced by ppTGRL, NT, LPS- and DEX-treated HMDM were incubated (24 h) with 0, 10 and 30 μg of ppTGRL CH/mL and the cellular lipid content was measured by fluorescent methods26,27 (Figure 4). In the absence of lipoprotein, the intracellular TG content of NT, LPS- and DEX-treated macrophages were similar (Figure 4a), and pp-TGRL induced dose-dependent TG accumulation in all the three cell types (RM-ANOVA, concentration, P < 0.002), with no significant differences between the treatments. When the individual ppTGRL concentrations were compared, however, the increment of TG content between macrophages incubated with 0 and 30 μg of CH/mL ppTGRL was significant only in DEX-treated group (253 ± 37 and 326 ± 30 nmol TG/mg cell protein; P < 0.05, n ≥ 3).
After incubation in 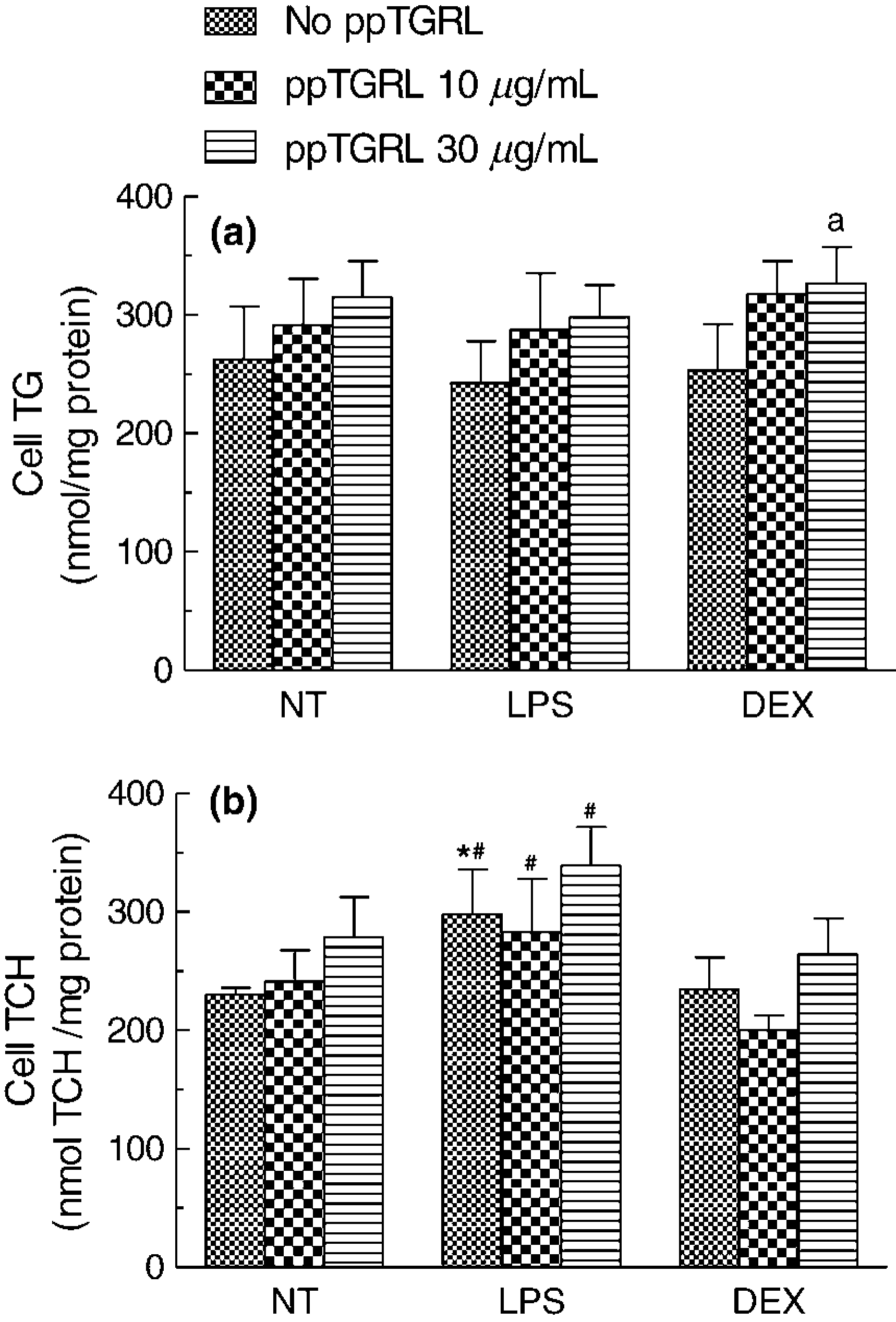
ppTGRL also increased macrophage CH levels in a dose-dependent way in NT macrophages (RM-ANOVA, P < 0.05), but in this case the inflammatory phenotype of macrophages had a significant effect, with CH concentrations being higher in LPS-treated as compared with DEX-treated cells (RM-ANOVA, treatment, P < 0.002) (Figure 4b). Furthermore, the dose-dependent effect of ppTGRL was lost in both LPS- and DEX-treated macrophages; and at any concentration of ppTGRL, the CH accumulation was significantly higher in LPS-treated as compared with DEX-treated macrophages.
Macrophage phenotype, ppTGRL and macrophage TG and PL synthesis
To evaluate whether the intracellular synthesis of TG and PL is modified by the macrophage M1 or M2 phenotype, NT, LPS- or DEX-treated HMDM were incubated with increasing concentrations (0-30 μg CH/mL) of ppTGRL in the presence of [3H]glycerol. In the absence of ppTGRL, [3H]glycerol incorporation into TG by the cells was similar irrespective of LPS or DEX treatment (Figure 5a). The presence of ppTGRL induced a dose-dependent increase in [3H]glycerol incorporation into TG in all three conditions (P < 0.001, RM-ANOVA). In NT cells, [3H]TG synthesis was significantly increased by 10 and 30 μg/mL ppTGRL (P < 0.001), however, in LPS- and DEX-treated cells the concentration-dependent increase was smaller and only the 30 μg/mL concentration caused a significant change (P < 0.001 for both groups). [3H]TG synthesis was significantly lower in DEX-treated than in NT macrophages at both concentrations of ppTGRL tested (10 μg/mL, P < 0.05; 30 μg/mL P < 0.01), but there was no significant change with LPS treatment (Figure 5a).
Incorporation of [3H]glycerol into cellular lipids were used to evaluate the synthesis of TG (a) and of phospholipids (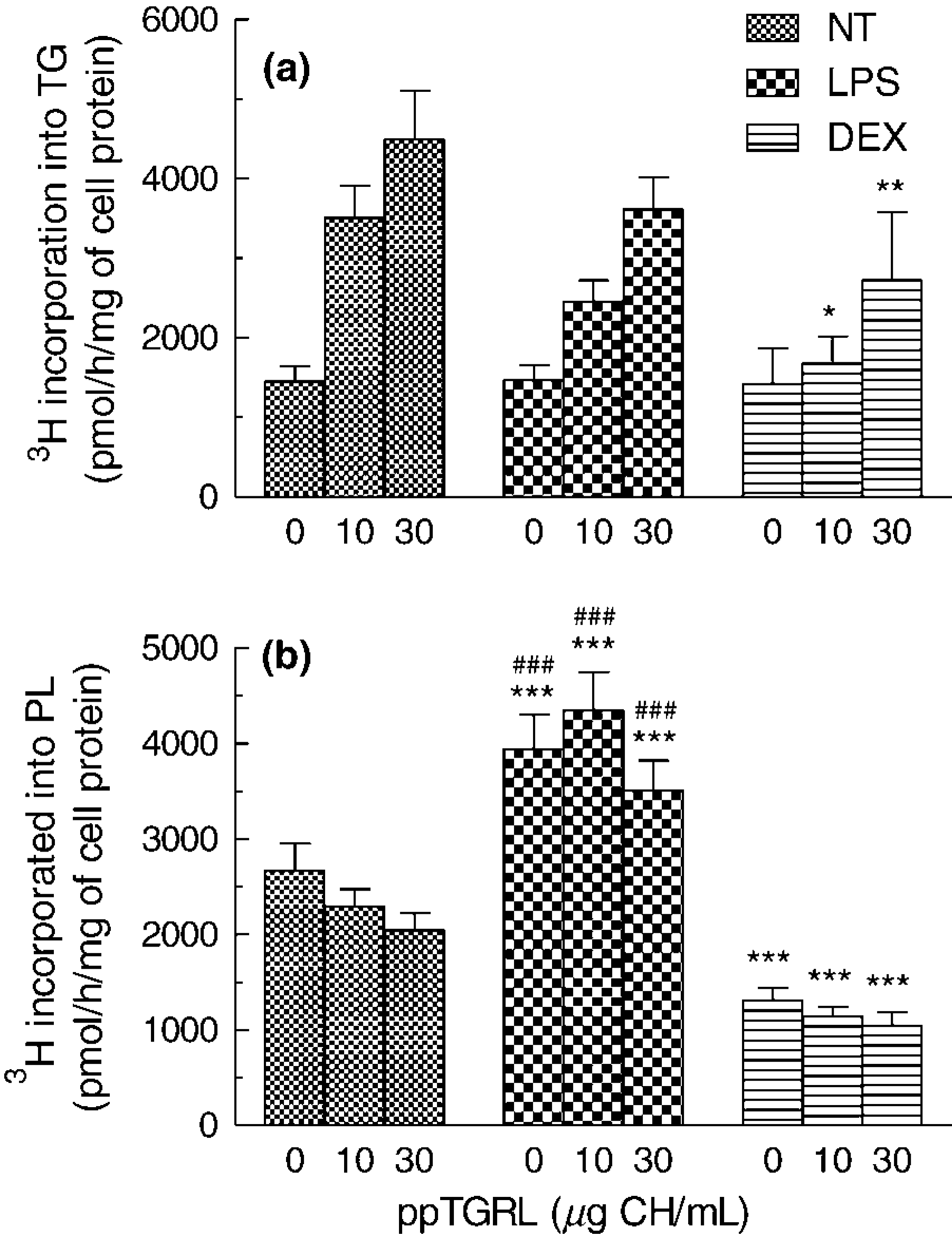
The main effects observed on PL synthesis were caused by the pro- and anti-inflammatory treatments (RM-ANOVA P < 0.0001), rather than addition of ppTGRL. Synthesis was strongly enhanced in LPS-treated as compared with NT cells, but markedly decreased in DEX-treated macrophages (P < 0.001) in the absence of added lipoprotein, and these differences were maintained at both ppTGRL concentrations tested. There was a dose-dependent decrease in [3H]glycerol incorporation into PL in the presence of ppTGRL in NT and DEX-treated cells (P < 0.05), although this did not quite reach significance in the former case (P = 0.06) (Figure 5b).
Since the amount of ppTGRL isolated from a single donor was sometimes insufficient to test the effects of numerous factors on the incorporation of [3H]glycerol into TG and PL, CRLP, which have been widely used to mimic chylomicron metabolism21,37–39 were used. Measurement of [H]glycerol incorporation into TG and PL in HMDM incubated with CRLP (30 μg CH/mL) gave similar results to those obtained with ppTGRL, with CRLP (30 μg CH/mL) inducing an increase in [3H]TG synthesis which was greater in NT (×3.6)- than in LPS (×2.1)- or DEX (× 2.8)-treated cells. In addition, [3H]PL synthesis by LPS stimulated cells was higher than in NT and DEX-treated macrophages (NT, 3436 ± 750; DEX, 2661 ± 780; LPS 4632 ± 1330 pmol/h/mg cell protein, P<0.05). Since the results obtained with both physiological and artificial TGRL were similar, the following experiments on [3H]glycerol incorporation into lipids were performed with either ppTGRL or CRLP and the results were pooled.
Effects of extracellular lipases activity on metabolism of ppTGRL in LPS- and DEX-treated macrophages
Lipases secreted by macrophages include LPL, endothelial lipase (EL), carboxyl ester lipase (CEL) and secretory phos-pholipase A2 (sPLA2) type-IIA and type-GV, and supply substrate for cellular TG and PL synthesis without internal-ization of whole lipoprotein.21,40–46 Initial experiments showed that in HMDM, LPL and CEL are the most highly expressed lipases. However, detectable, but low levels of expression, in decreasing order, of sPLA2 GV, sPLA 2IIA and EL were also found (data not shown).
To explore the hypothesis that LPS and DEX affect TG and PL synthesis in the presence of ppTGRL (Figure 5) through the regulation of macrophage lipolytic activities, HMDM were incubated with 30μg CH/mL of ppTGRL together with Orlistat or way121.989 (way), which are known inhibitors of LPL and CEL,36,37 respectively. Lipid synthesis was evaluated by [3H]glycerol incorporation into TG and PL. The results (Figure 6) showed that Orlistat caused a strong inhibition of [3H]TG synthesis in NT, LPS-and DEX-treated cells (71%, 53% and 70%, respectively) (Figure 6a). However, there was significantly less inhibition in LPS-activated cells compared with NT (P < 0.05, n = 4) and DEX-treated HMDM. The CEL inhibitor, way, consistently reduced the production of [3H]TG by about 20% in the NT, LPS- and DEX-treated groups, but these changes did not reach significance. In comparison with control cells, Orlistat and way did not change the incorporation of [3H]glycerol into [3H]PL (Figure 6b), with the exception that PL synthesis was decreased by Orlistat in LPS-treated (-27% of control cells) as compared with DEX-treated (+8% of control cells) macrophages (P < 0.05).
Effects of secretory macrophage lipase inhibition on intracellular synthesis of TG (a) and of phospholipids (PL) (b), induced by ppTGRL in 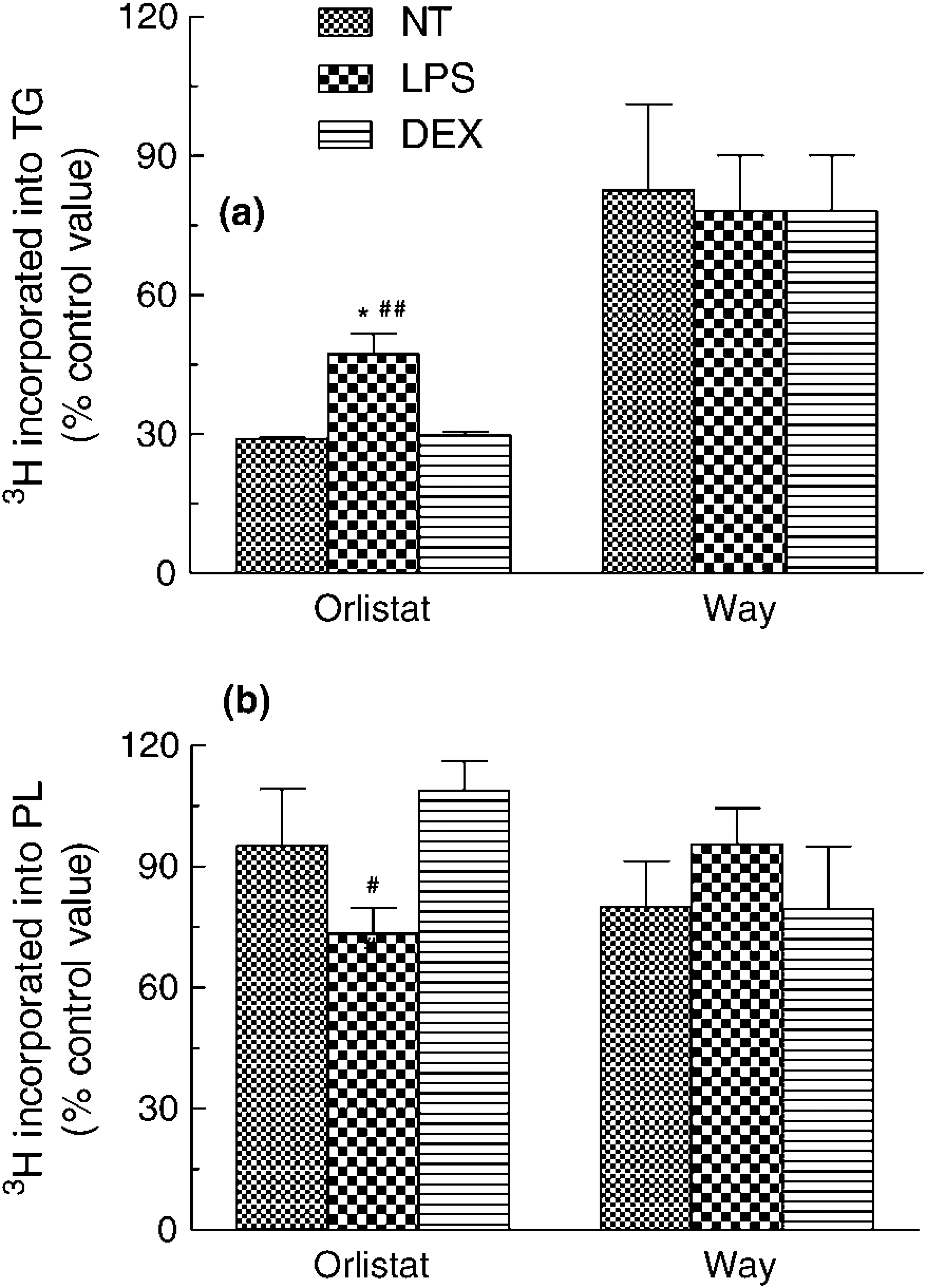
Effect of macrophage phenotype on secretory lipase expression and activity
We next investigated whether and how the expression of mRNA for LPL and CEL is modulated by LPS and DEX treatments (Figure 7). With respect to NT cells, LPS decreased the levels of both LPL (P < 0.05, n = 5) (Figure 7a) and CEL (Figure 7b) mRNA by approximately 50%, while DEX caused a significant increase in mRNA for both LPL (P< 0.01, n = 5) and CEL (P < 0.01, n = 3) of about 2.5- and 7-fold, respectively.
HMDM were treated for 18h in complete 
The activity of LPL secreted by NT, LPS- and DEX-treated macrophages is shown in Figure 7c. In DEX-treated cells, the released LPL activity (41 ± 1.9 nmol fatty acids/h/mL) was significantly higher than in LPS-treated (18.1 ± 1.2 nmol fatty acids/h/mL, P < 0.01) and NT macrophages (25 ± 1.5 nmol fatty acids/h/mL, P < 0.05) cells, confirming that LPL activity was down-regulated in the LPS-activated state and up-regulated in the anti-inflammatory state induced by DEX.
Macrophage phenotype and CH metabolism
To test the hypothesis that LPS and DEX modulate the metabolism of CE carried by ppTGRL by regulating the selective uptake of CE, we incubating LPS- and DEX-treated macrophages with [3H]cholesteryl oleyl ether-([3H]Cether) or [3H]CE-labeled CRLP. Cether is taken up but not released by macrophages and thus cell [3H]Cether is a measure of CE internalization. In contrast, CE delivered in lipoprotein to macrophages is hydrolysed, allowing UC mobilization from cells to extracellular acceptors (reverse CH transport: RTC). 47 In the experiments with [3H]CE-CRLP, therefore, the total cell radioactivity (UC + CE) reflects the balance between CE internalization and CH efflux. Incorporation of [3H]Cether-CRLP (Figure 8a) into HMDM was similar in pro-activated and anti-inflammatory macrophages, suggesting that the differences in the induction of CH accumulation by ppTGRL in cells in the two macrophage phenotypes (Figure 3) are not due to an effect on the selective uptake of CE.
HMDM were pretreated for 18h in complete 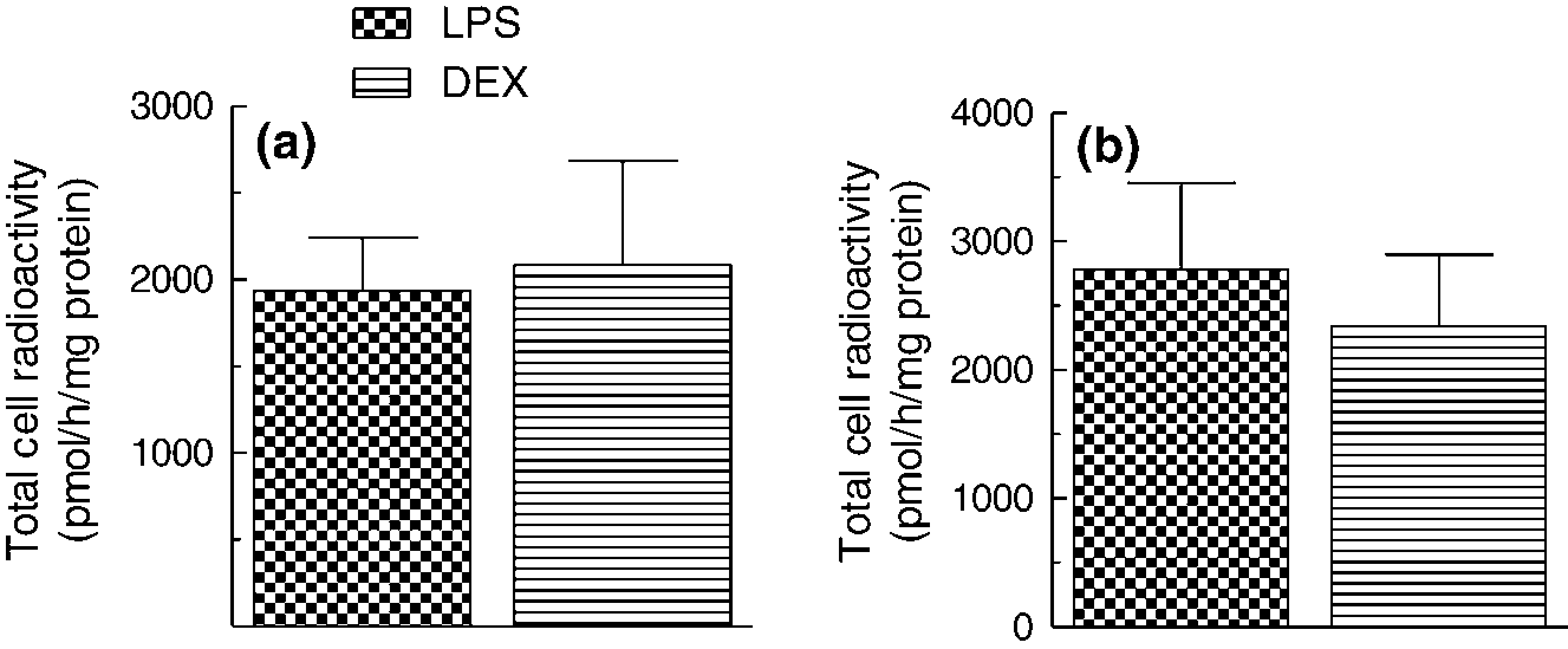
In macrophages incubated with [3H]CE-CRLP (Figure 8b) the radioactivity recovered tended to be higher in LPS-treated than in DEX-treated cells, and the difference almost reached significance (paired Student's t-test, P = 0.06, n = 3). Thus, since no differences were found in the experiments with [3H]Cether, the change in [3H]CE incorporation may be due to a difference in the rate of removal of CH from activated M1 as compared with anti-inflammatory M2 cells. Block lipid transport (BLT) inhibitors are small molecules that inhibit transporters mediating CH efflux. 47 The effects of inhibition of CH efflux from HMDM on the accumulation of radioactivity from [3H]CE-CRLP were tested using BLT-1, a specific inhibitor of SR-B1. 47 The recovery of radioactivity in the cells in LPS- and DEX-treated cells was found to be similar in the presence or absence of the inhibitor (data not shown). In another set of experiments, BLT-4, which blocks efflux via SR-B1 and the ATP-binding cassette transporter A1 (ABCA1) 47 was used (Figure 9). The addition of ppTGRL to the incubations causes a significant increment of cell CH in LPS-treated cells, but not in DEX-treated cells (250 ± 19 and 65 ± 22 nmol/mg cell protein, respectively; P < 0.05, n = 3) (Figure 9), but this difference was still observed in the presence of BLT-4, suggesting that the impairment of CH efflux is not a determining factor in the higher increment of CH accumulated by LPS macrophages with respect to DEX cells. BLT-4 did not alter the cellular CH content of cells exposed to LPS or DEX in the absence of lipoprotein (data not shown).
LPS- and 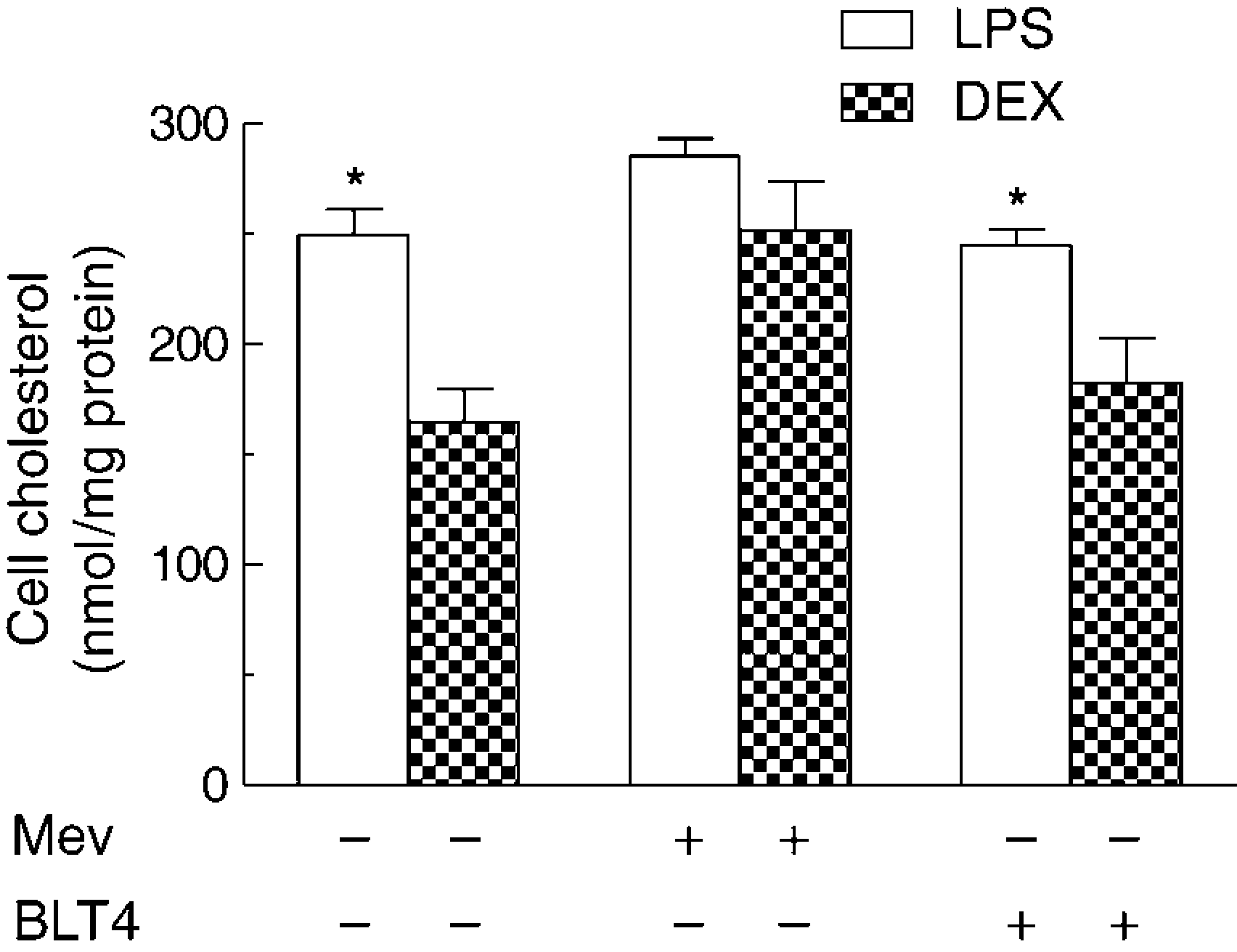
Feingold et al. 48 have found that endotoxin up-regulates hydroxyl-methylglutaryl-coenzyme A reductase (HMG CoA red) activity and thus causes increased CH synthesis, which could result in increased cellular CH levels. We investigated this possible explanation for the differential effects of ppTGRL on CH accumulation in LPS-challenged as compared with DEX-challenged macrophages using the HMG CoA red inhibitor, mevinolin. 49 In the presence of the inhibitor, the enhancement in CH accumulation in LPS-treated as compared with DEX-treated cells was abolished (Figure 9). These results suggest that CH synthesis is enhanced in pro-inflammatory M1 macrophages in the presence of ppTGRL.
Discussion
The specific aim of this study was to investigate the effect of ppTGRL on lipid metabolism in pro-inflammatory (M1) and anti-inflammatory (M2) macrophages induced by LPS and DEX treatment, respectively. To validate the investigation, however, our initial objective was to verify that LPS and DEX promote a shift towards the M1 or M2 phenotype in HMDM.
Assessment of our cellular model demonstrates that our population of mature HMDM expresses CD14 and CD68 (Figures 1 and 2) and is responsive to LPS and DEX, with the two types of challenge inducing differential patterns of antigen expression and cytokine secretion which are consistent with previous reports of the characteristics of the classically activated M1 and alternatively activated M2 phenotypes, respectively.34,35 It should be stressed that macrophages exist in a continuum of activation states of which the M1 and M2 phenotypes are the extremes. 50 Thus, various treatments may be expected to shift the balance away from one and towards the other, but not necessarily to produce all the effects associated with the extreme states. For example, secretion of IL-10, an anti-inflammatory cytokine, usually associated with the M2 phenotype 51 was higher after LPS as compared with DEX-treatment; however, in NT macrophages, the ratio between the secretion of the pro-inflammatory TNF-α and IL-10 was 1.0, and activation by LPS was shown to shift the balance towards a more inflammatory profile (TNF-α/IL-10 ratio: 1.3). In contrast, treatment with DEX increased secretion of both cytokines, with the production of IL-10 being higher than that of TNF-α, resulting in an anti-inflammatory metabolic profile (TNF-α/IL-10 ratio: 0.74). Thus, although the and LPS- and DEX-treated HMDM did not show all the features of M1 and M2 macrophages, our findings indicate that the balance is shifted towards a more inflamed M1 pheno-type after LPS treatment and to a more anti-inflammatory M2 profile when DEX is used. This model, therefore, is suitable for the investigation of the interaction between inflammation and the effects of ppTGRL on macrophage lipid metabolism.
For most experiments in this study, we used the d = 1.006 g/mL lipoprotein fraction isolated from the plasma of normolipidemic subjects three hours after consumption of a standard fat-rich breakfast. This fraction is rich in chylomicron remnants, but also contains other TGRL, such as VLDL and VLDL remnants. 36 However, for some experiments where it was not possible to obtain sufficient ppTGRL, CRLP, 22 which resemble physiological chylomicron remnants in their size, density and lipid composition, and mimic their effects in cultured cells in vitro52–54 were used. These particles have the advantage that their composition is easily reproducible, but the drawback that they do not contain apoB48 and do not show the heterogeneity which is a major feature of physiological ppTGRL.
The TG/CH molar ratio of ppTGRL used in this study (Table 2) was similar to that reported previously, 6,36 and the fatty acid composition reflected that test meal (Table 2) as would be expected. 55
Vessel wall-resident macrophages undergo local activation in response to various inflammatory and immune stimuli which modulate their phenotype and induce activation 14 and this, in turn, plays a key role in foam cell formation.1–3 The role of macrophage activation in chylomicron remnant metabolism has not been addressed previously, but studies with LDL have show that lipid accumulation depends on both the pro-inflammatory activation induction factor and the type of LDL used.15–18,56–58 In addition, macrophage activation by LPS has been found to inhibit scavenger receptor activity, reducing the uptake of modified LDL,15,57–60 and to increase lipid accumulation induced by native LDL, either via macropino-cyosis 16 or induction of LDL receptor expression. 18 Assessment the effect of LPS on LDLr and scavenger receptor activities in HMDM our experimental conditions, however, showed no significant change in the activity of the LDLr or scavenger receptors in LPS-treated cells.
Lipid accumulation and synthesis
In previous work, ppTGRL, 41 TGRL obtained from hyper-triglyceridemic subjects 60 and CRLP21,24,39 have been shown to induce macrophage foam cell formation. Furthermore, after activation of macrophages by exposure to LPS or phorbol ester, foam cell formation can be induced by native LDL.15–17 Little attention, however, has been paid to the effects of macrophage inflammatory phenotype on their response to TGRL, although Funk et al. 15 have reported that LPS increases the induction of CH accumulation by β-VLDL in a murine macrophage cell line. Here, we found that TG accumulation was increased by ppTGRL in a dose-dependent way, regardless of the inflammatory phenotype of HMDM (Figure 4a). In contrast, cell CH levels were increased by LPS, but not DEX treatment in the absence and presence of ppTGRL, and the ppTGRL dose-dependence was lost in both cases. These results suggest that inflammation promotes lipid accumulation in macro-phages, that this effect overrides the smaller effect of ppTGRL, and is related to the cellular handling of CH rather that TG.
The amount of CH that accumulates within cells is clearly a balance between that taken up or synthesized and that removed by RCT. Thus, the rise in intracellular CH levels which occurs when HMDM are polarized towards the M1 phenotype may be caused by increased CH entry and/or synthesis and /or decreased efflux. CH from lipoproteins may enter the cells by uptake of whole particles via receptors such as the LDLr, or by selective uptake of CE. 21 As indicated above, we were unable to detect any change in the LDLr or scavenger receptor activity caused by LPS in our experiments with HMDM, and we found no evidence to suggest that the selective uptake of CE from these particles was changed by LPS-treatment (Figure 6).
As CH synthesis in the liver has been shown to be up-regulated by endotoxin via increased activity of 3-hydroxyl-methylglutaryl-Coenzyme A reductase, 48 we investigated the effects of inhibition of the enzyme with mevinolin in HMDM. The inhibitor was found to abolish the difference between CH accumulation in LPS- and DEX-treated cells in the presence of ppTGRL, suggesting that CH synthesis is enhanced in macrophages expressing a more inflammatory phenotype (Figure 9).
LPS has also been found to down-regulate the expression in macrophages of SR-B1 and ABCA1, two proteins which play a major role in the removal of CH from the cells.59–61 Our studies using inhibitors of SR-B1 and ABCA1 (BLT-4) (Figure 9), and of SR-B1 (BLT-1) (data not shown), indicate that the effects of LPS in increasing CH accumulation in the presence of ppTGRL are not due to decreased CH efflux. Furthermore, although it has been reported that the LDL receptor-related protein (LRP) has an important role in CRLP uptake by the monocyte–macrophage cell line, THP-1, 12 we excluded that the present results could be attributed to a modulation of this receptor, as our attempts to regulate the LPS/DEX effects with lactoferrin, a well-known inhibitor of LRP function, did not affect the results (data not shown). Thus, the increased induction of CH accumulation in inflammatory as compared with anti-inflammatory HMDM cannot be accounted for by increased entry via the LDLr or scavenger receptors, selective CE uptake or decreased CH efflux, but may be due to a rise in the rate of CH synthesis. However, as we found a strong effect of macrophage activation on PL metabolism, we cannot rule out the possibility that the changes in cell lipid content may be related to changes in the composition of membrane domains which are not measured by these methods. 62
Lipases and ppTGRL
Our previous work has shown that chylomicron remnants cause increased synthesis of TG in HMDM and murine J774 macrophages21,24 and others 41 have reported a similar effect with TGRL. In the present study using HMDM, ppTGRL were also found to increase TG synthesis. This induction, however, was unaffected by increased inflammation, but was dampened in anti-inflammatory conditions (Figure 5a). Independently of ppTGRL, LPS caused a marked rise in PL synthesis, while DEX caused a fall of a similar magnitude (Figure 5b), suggesting that macrophage PL synthesis/ turnover increases with increasing inflammatory phenotype, and this effect is not dependent on the presence of lipoproteins.
In our previous work, inhibition of LPL activity in the presence of chylomicron remnants was found to cause decreased cellular TG levels and increased TG synthesis, suggesting that fatty acids released extracellularly by LPL are taken up by the cells and used to synthesize TG. 21 Evaluation of the HMDM secretory lipases in the present study indicated that LPL and CEL were the most strongly expressed, although lower levels of sPLA2 GV, sPLA 2IIA and EL were also detected. In agreement with our earlier findings, 21 inhibition of LPL activity by orlistat caused a marked decrease in TG synthesis in HMDM treated with ppTGRL, while the CEL inhibitor way had no significant effect (Figure 6a), suggesting that CEL does not play an important role in the uptake of ppTGRL lipid by the cells. These experiments also show that TG synthesis remains significantly higher in pro-inflammatory as compared with anti-inflammatory cells even when LPL activity is inhibited. Moreover, inhibition of extracellular LPL markedly reduced the esterification of fatty acids to PL in LPS-challenged, but not untreated or DEX-treated, cells, indicating that in the activated state the fatty acids delivered from extracellular lipolysis are preferentially driven towards PL rather than TG synthesis.
Further investigation showed that LPL and CEL mRNA expression was up-regulated in anti-inflammatory macrophages and, in agreement with previous reports using other macrophage types,43,63 down-regulated in pro-inflammatory cells (Figure 7a, b). The activity of LPL followed a similar pattern to mRNA expression, being strongly up-regulated by DEX treatment and down-regulated by LPS (Figure 7c). We conclude, therefore, that the raised TG synthesis induced by ppTGRL in pro-inflammatory as compared with anti-inflammatory macrophages is not due to enhanced LPL activity causing increased release of fatty acids extracellularly. This conclusion is further supported by the finding that in NT and DEX-treated macrophages, LPL inhibition reduced the synthesis of TG more markedly than in the LPS-treated group (Figure 6). In addition, these data indicate that the changes in PL synthesis induced by LPS and DEX in the absence of lipoproteins are not related to changes in extracellular LPL activity, but rather to the effects of macrophage inflammatory phenotype on the intracellular metabolism of PL. However, since the incorporation of [ 3 H]glycerol into PL is inhibited by the presence of orlistat, the lipolytic activity may be important in the rise in PL turnover induced by endotoxin treatment (Figure 5b).
The modulation of macrophage inflammation by DEX and LPS may have, in fact, an important function in atherosclerosis progression. In subclinical carotid stenosis, subjects with echolucent carotid plaques have been shown to have decreased postheparin LPL activity accompanied by delayed clearance of ppTGRL, 63 which would cause a prolonged pp-triglyceridemia, and an unfavorable translo-cation of cholesteryl-esters from HDL to chylomicron remnants, 64 increasing the risk of atherosclerosis. In addition, the decrease in LPL activity caused by inflammation may contribute to the promotion of atherogenesis through other mechanisms, mainly related to levels of lysophos-phatidylcholine.65,66 Thus, the present results show that the regulation of the expression and activity of LPL by pro- and anti-inflammatory factors play a key role in determining macrophage lipid accumulation in the pp phase.
In summary, in this study primary human macrophages polarized towards a classically activated M1 or an alternatively activated M2 phenotype by treatment with LPS or DEX were used, together with ppTGRL derived from human volunteers, to investigate the interactions between inflammation and the effects of ppTGRL on lipid metabolism in the cells. The results indicate that inflammatory M1 as compared with anti-inflammatory M2 macrophages accumulate more CH, and that the effect of inflammation is greater than that of ppTGRL. We found evidence to suggest that the higher CH content of LPS-treated HMDM may be caused by increased CH synthesis, but appears not to be directly linked to an increase in neutral lipid uptake from ppTGRL, or a decrease in the rate of CH removal from the cells. In the presence or absence of lipo-proteins, PL synthesis is markedly increased in inflammatory as compared with anti-inflammatory HMDM, and a higher proportion of fatty acids resulting from extracellular lipolysis by LPL are used for PL, rather than TG, formation. In addition, ppTGRL were found to cause an increase in the rate of TG synthesis in HMDM which is greater in inflammatory cells, and this effect is not due to changes in LPL activity. We conclude, therefore, that the neutral lipid accumulation that occurs in pro- inflammatory M1 as compared with anti-inflammatory M2 macrophages in the presence or absence of ppTGRL is related to changes in PL metabolism and possibly also CH synthesis. Overall, these findings indicate that lipid metabolism is altered by the activation state of macrophages, and this influences the metabolism of TGRL in the postprandial phase, leading to increased foam cell formation.
Footnotes
Acknowledgements
We are grateful to Massimo Sanchez for his professional help in macrophage phenotyping. This work was supported by grants from the Istituto Superiore di Sanita’ (Rif. ISS: 30F6) and by the Ricerca Finalizzata of the Italian Ministry of Health (Ref ISS R26).