
Abstract
The objective of the present paper is to develop and apply a multiplex polymerase chain reaction (mPCR) based reverse line blot (RLB) hybridization assay to facilitate the diagnosis of genital infections by detection of seven recognized or putative genital pathogens (Neisseria gonorrhoeae, Chlamydia trachomatis, Ureaplasma urealyticum, Ureaplasma parvum, Mycoplasma genitalium, Mycoplasma hominis and Trichomonas vaginalis). Species-specific biotin-labelled primer pairs were used in a single mPCR to amplify target regions in each of seven pathogens. The amplified biotin-labelled PCR products were hybridized with membrane-bound-specific oligonucleotide probes and were detected by chemiluminescence. Two hundred and eleven specimens (104 male urethral and 107 female vaginal swabs), collected from patients with suspected genital infections attending the Wuhan First Hospital Sexually Transmitted Diseases (STD) clinic, were tested by mPCR/RLB and results were confirmed by single PCR using different species-specific targets. The sensitivity of the assay was assessed using dilutions of positive DNA controls with known copy numbers, for each target. The assay correctly identified all reference strains and detected potential pathogens in a high proportion of clinical specimens. There was no cross-reaction between the seven pathogens. The mPCR/RLB can detect ≤ 102 copies of the target gene fragments. Comparison of mPCR/RLB and single PCR assays showed discrepant results in six of 211 (2.8%) clinical specimens, which were positive by mPCR/RLB assay, but negative by the corresponding single PCR. Nested PCR on the six discrepant specimens gave results consistent with those of mPCR/RLB. In conclusion, the mPCR/RLB hybridization assay is sensitive and specific, and able to rapidly detect genital pathogens in clinical specimens.
INTRODUCTION
Sexually transmitted infections (STIs) are an important public health problem worldwide. In men, urethritis, characterized by dysuria and urethral discharge, is among the most common manifestations, with an estimated global incidence of ∼150 million cases annually. 1 In women, vaginal discharge resulting from cervicitis, vaginitis or bacterial vaginosis is common. Both syndromes can be caused by one of several well-recognized pathogens but the cause is often uncertain or unknown.
Recognized sexually transmissible pathogens include Neisseria gonorrhoeae and Chlamydia trachomatis. The World Health Organization (WHO) estimates that 62 million cases of gonorrhoea, many of which present as urethritis, occur annually worldwide. 2 C. trachomatis is the major cause of non-gonococcal urethritis (NGU), accounting for 30–50% of cases. 3 Both organisms can also infect the cervix, urethra, oropharynx, conjunctiva and rectum, and can cause pelvic inflammatory disease (PID) and its complications in women. 4
Genital mycoplasmas have been implicated in NGU, bacterial vaginosis and PID, but their roles are less well-defined 5 in part because they are frequently found among the genital flora of healthy men and women. Human ureaplasmas have been recently divided into two species – Ureaplasma urealyticum and Ureaplasma parvum 6 – the respective pathogenic roles of which remain controversial.
Mycoplasma genitalium is found uncommonly in asymptomatic men and women (1–2%) 7 but can be detected in 12–14% of men with non-chlamydial NGU, 8 and there is also evidence of its involvement in female genital infections including PID. 9 Mycoplasma hominis is associated with bacterial vaginosis, PID and possibly urethritis. 10
Trichomonas vaginalis is a recognized cause of vaginitis. It is infrequently implicated in NGU but may be carried asymptomatically and transmitted to sexual partners; it has been implicated as a co-factor in the transmission of HIV. 11
Culture has been the ‘gold standard’ for diagnosis of many STIs, but increasingly has been replaced, especially for slowly growing or non-culturable pathogens, by antigen or nucleic acid detection tests. The use of reverse line blot (RLB) hybridization can enhance postamplification detection of multiplex PCR (mPCR) products and allows rapid, simultaneous detection of several pathogens with high sensitivity and specificity. 12 Our aim was to develop and evaluate an mPCR-based RLB assay to facilitate the diagnosis and epidemiological investigation of STI.
MATERIALS AND METHODS
Reference strains
C. trachomatis serotype H ATCC UR-88, M. genitalium ATCC 33530, Mycoplasma fermentans ATCC 19989, M. pneumoniae ATCC 29342, 14 serovars of U. parvum and U. urealyticum reference strains were obtained directly from the American Type Culture Collection (ATCC). T. vaginalis liquid culture was provided by Dr Jacqui Upcroft of the Queensland Institute of Medical research. Clinical isolates of N. gonorrhoeae, M. hominis, N. meningitidis and Chlamydia pneumoniae strains were used as reference strains.
Clinical specimens and DNA extraction
Urethral swabs were collected from 104 consecutive male patients with symptoms of urethritis and vaginal swabs from 107 consecutive female patients with vaginal discharge, attending the Wuhan First Hospital STD clinic (Wuhan, PR China).
DNA was extracted as described previously. 13 Briefly, swab tips were broken off into 2 mL of PCR digestion buffer (10 mmol/L Tris-HCl, pH 8.0, 0.45% Triton X-100 and 0.45% Tween 20), which was then vortexed for 20 s and centrifuged at 4800 rpm for 30 min at room temperature. The pellet was resuspended in 200 µL of PCR digestion buffer and 2 µL of proteinase K (20 mg/mL), mixed, incubated at 55°C for one h and heated to 95°C for 20 min. Extracts were stored at −20°C until tested.
mPCR/RLB primer and probe design
16S–23S rRNA intergenic spacer sequences for Neisseria, Chlamydia, Ureaplasma and Mycoplasma spp. were compared, using ClustalW program in Biomanager (
Primers and probes used in multiplex polymerase chain reaction (mPCR)/reverse line blot (RLB)
*‘S’ indicates ‘sense’; ‘A’ indicates ‘antisense’ primers
†‘b’ indicates 5′ biotin-labelled primer
‡‘p’ indicates 5′amino-labelled probe
Multiplex PCR amplification
The seven primer-pair mPCR mixture was prepared as follows: 5 µL template DNA, 0.25 µL of all forward primers (50 pmol/µL) and reverse primers (50 pmol/µL), 1.25 µL dNTPs (2.5 mmol/L of each dNTP), 2.5 µL 10 × PCR buffer, 3 µL 25 mmol/L (4.5 mmol/L final) MgCl2, 0.3 µL QIAGEN HotStar Taq polymerase (5 units/µL) (QIAGEN, Pty Ltd, Doncaster, Australia), with water added to 25 µL. Multiplex PCR was performed according to instructions from QIAGEN HotStar Taq polymerase suppliers as follows: 95°C for 15 min, one cycle; 94°C for 30 s; 55°C for 30 s; 72°C for one min, 35 cycles; 72°C for 10 min, one cycle; and 22°C hold. A measure of 15 µL of PCR product was used for RLB hybridization. DNA extracts from reference strains were used as controls.
Reverse line blot hybridization
The RLB hybridization assay was based on the method described previously, 12 except that the hybridization temperature was 60°C, streptavidin-peroxidase conjugate (Roche Diagnostics Co, Mannheim, Germany) was diluted 1:4000 in 2 × SSPE (1 × SSPE is 0.18 mol/L NaCl, 10 mmol/L NaH2PO4, 1 mmol/L EDTA [pH 7.7])–0.5% sodium dodecyl sulphate, and the time of exposure of the X-ray film (Hyperfilm; Amersham, Buckinghamshire, UK) was adjusted to seven min. To optimize hybridization conditions, the probe was tested at several two-fold dilutions as previously described. 13
Plasmid construction and standard template
Each reference strain, excect T. vaginalis, was amplified using 16S-23S rRNA intergenic spacer region ‘universal primers’, 16S1 and 23A1 (Table 1). T. vaginalis repeat DNA fragment was amplified using primers TVK3 and TVK7 (Table 1). Amplified products were inserted into pGEM-T vector using pGEM-T Easy Vector Systems (Promega, Madison, WI, USA) and transformed into competent Escherichia coli JM109 cells, according to the manufacturer's protocol.
15
Screening for the gene insert was performed by quick lysis at 95ºC of the E. coli cells for five min, followed by PCR, using pGEM-T primer and 5 µL of the cell lysate. All PCR products were purified and sequenced using the Big-Dye Terminator Cycle Sequencing Ready Reaction kit (ABI Prism, v 3.0, Foster City, California, USA) on an ABI 377 automated sequencer. Amplified products, including target DNA confirmed by sequencing, were used as standard templates. The copy numbers in each were estimated by A
260 measurement (Biophotometer, Eppendorf, Germany) to estimate the total molecular weight of the amplified product and copy number was calculated using the formula provided by the website of URI Genomics & Sequencing Center (
mPCR/RLB sensitivity
To estimate the sensitivity of the assay, reference strain standard templates were serially diluted from 104 to 102 copies per reaction and tested by mPCR/RLB.
Detection of mixed DNA
N. gonorrhoeae, C. trachomatis and U. urealyticum standard templates were mixed, so that each was present in different proportions – 1%, 5% or 10% of a total of 105 copies – in separate mPCR reactions. PCR products were detected by RLB.
Single species-specific PCRs to confirm mPCR/RLB results
Previously published species-specific primers and amplification conditions for each target species were selected (Table 2). Nested PCR was used for further confirmation if results of mPCR and single PCR differed. The outer primers were the ‘universal’ 16S–23S rRNA intergenic sequence primers (16S1 and 23A1), as described above (Table 1) and the inner primers were another ‘universal’ forward inner primer (16S2) and the species-specific antisense probes.
Polymerase chain reaction (PCR) primers used for single species-specific PCR and nested PCR
RESULTS
mPCR/RLB specificity
All reference strains were correctly identified by mPCR/RLB. There were no cross-reactions between the seven target species (Figure 1) or with related species N. meningitidis and C. pneumoniae.
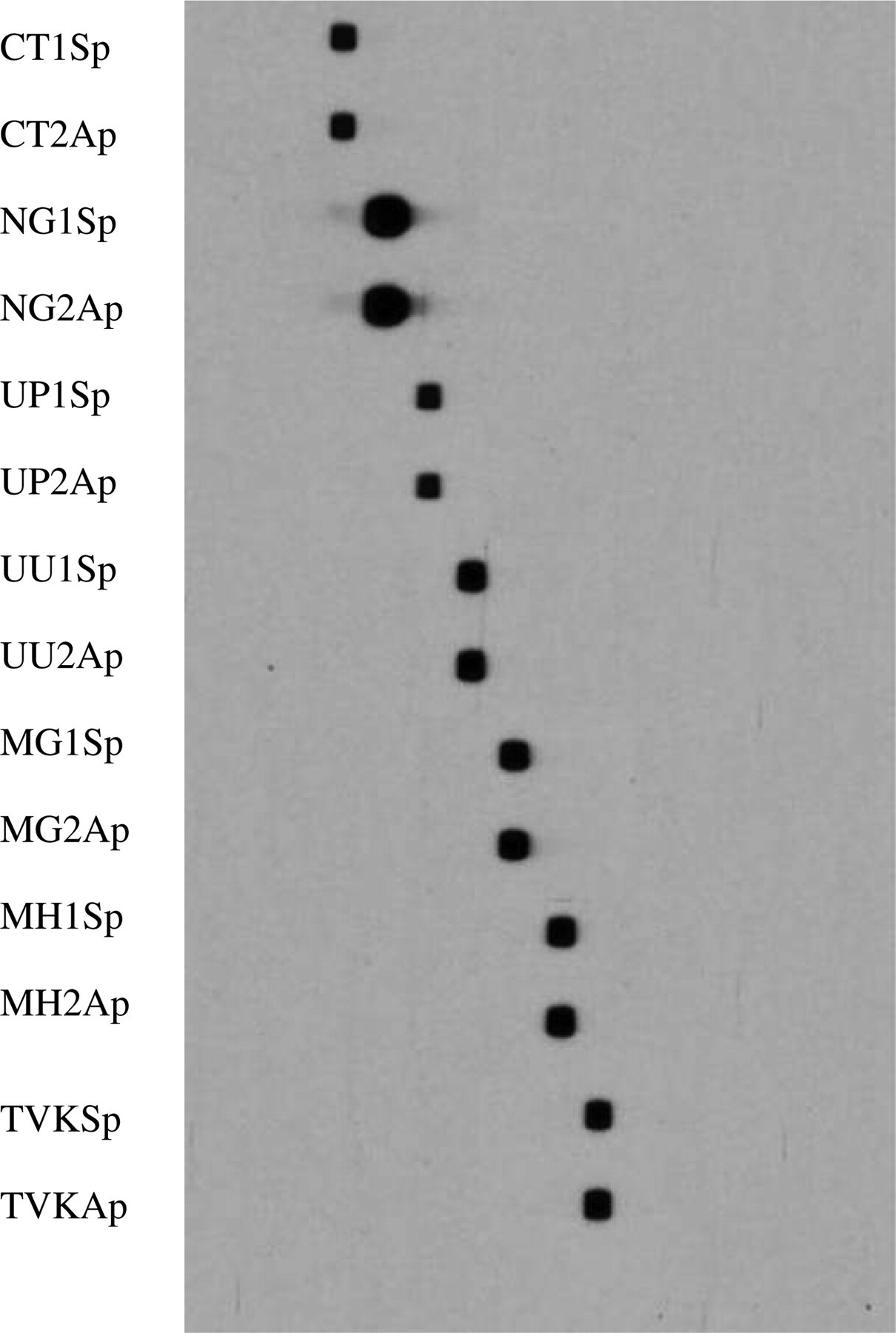
Seven reference strains of target species identified by multiplex polymerase chain reaction/reverse line blot method. The positions of 14 species-specific probes (two for each strain) are shown on the left. 1. Chlamydia trachomatis (H) ATCC 23206; 2. Neisseria gonorrhoeae, a well-characterized clinical isolate; 3. Ureaplasma parvum, ATCC 27618; 4. U. urealyticum, ATCC 27618; 5. Mycoplasma genitalium, ATCC 33530; 6. M. hominis, a well-characterized clinical isolate; 7. Trichomonas vaginalis liquid culture provided by Dr Jacqui Upcroft of Queensland Institute of Medical research (QIMR)
mPCR/RLB sensitivity
Dilutions of template DNA from each of the seven target species were tested by mPCR/RLB. All seven were detectable at the lowest copy number (102) tested (Figure 2).
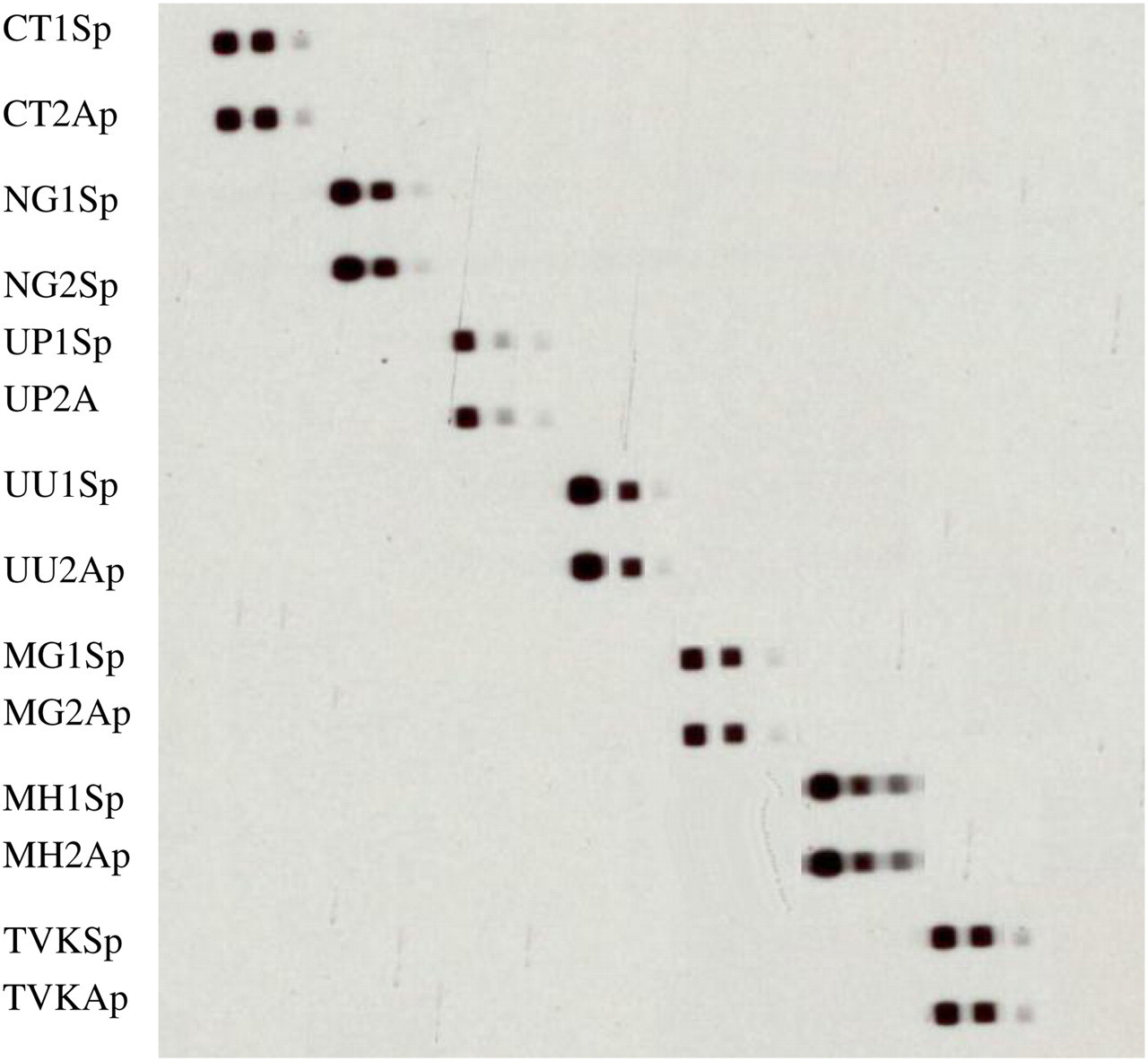
Multiplex polymerase chain reaction/reverse line blot sensitivity. Template DNA for each species, with known copy numbers, was diluted to produce specimens for sensitivity testing with known copy numbers. Results for dilutions containing 104, 103 and 102 DNA copies are shown from left to right for each species. All species were detectable at 102 copies
Mixed DNA detection
Mixtures of N. gonorrhoeae, C. trachomatis and U. urealyticum DNA templates, in differing proportions (1%, 5% or 10%), were tested by mPCR/RLB. N. gonorrhoeae and C. trachomatis templates were each detectable at a concentration of 1% and U. urealyticum template at 5% (data not shown).
Testing clinical specimens
One or more potential pathogens was detected in 106 of 211 (50%) clinical specimens, including 72 with one, 30 with two and four with three target species detected (total 144 target organisms detected). N. gonorrhoeae (in 21 specimens) and U. urealyticum (18) were the most commonly detected species in men and U. parvum (23) and M. hominis (18) in women (Table 3).
Comparison of results of multiplex polymerase chain reaction reverse line blot (mPCR RLB) and single PCR in 106 of 211 clinical specimens* (data are total number of specimens in which each species was detected by each method)
*No target organisms were identified in 105 of 211 specimens; a single organism in 72; two in 30 and three organisms in four specimens (total 144)
†There were six discrepant results between mPCR/RLB and single PCR
Single PCRs were performed for each species in all 211 specimens (138 individual PCR) and the results compared with mPCR/RLB. There were six discrepant results, all of which were positive by mPCR/RLB and negative by single PCR. Species identified were N. gonorrhoeae in three specimens and C. trachomatis, U. urealyticum and M. hominis in one specimen each. Nested PCR confirmed the mPCR/RLB result in all cases (Table 4).
Single and mixed species identified by multiplex polymerase chain reaction/reverse line blot (RLB) in 106 clinical specimens (of 211 tested) with positive results identified RLB result
DISCUSSION
We have previously used mPCR/RLB to detect several bacterial pathogens, simultaneously, in human respiratory 16 or blood 17 specimens using different species-specific targets, and to identify 10 mollicutes (mainly Mycoplasma and Ureaplasma spp.) in cell culture 13 using ‘universal’ primers targeting 16S–23S rRNA. These assays are highly sensitive and specific. The genital pathogens targeted in this study were several unrelated bacterial species, including mollicutes and a protozoan, which made the use of a ‘universal’ target difficult. Therefore we designed seven species-specific primers and two probes for each pathogen. The targets for six of these pathogens were species-specific sequences in the 16S–23S rRNA intergenic spacer region, which harbours both conserved and variable domains, in bacteria. 18 We confirmed the specificity of the mPCR/RLB assay using reference strains, including some closely related species (Figure 1). All target species were amplified by the corresponding species-specific primers and the amplicons hybridized to both corresponding probes on the RLB membrane (and no others). The sensitivity was measured using dilutions of cloned templates. The assay detected ≦102 copies of template for each species, which is at least equivalent to the sensitivity of single-target PCR.
The sensitivity and specificity was further evaluated using clinical specimens from symptomatic patients attending an STD clinic. N. gonorrhoeae was identified in 28 specimens, by mPCR/RLB and in 25 using a single PCR targeting cppB, which is the more commonly used target. 19 Nested PCR, also targeting the 16S–23S rRNA spacer region, but using an N. gonorrhoeae species-specific probe as the inner primer, confirmed the mPCR/RLB detection results. These results are consistent with those of a recent multicentre study, which showed that 6% of N. gonorrhoeae isolates lack cppB, which suggests that the maximum achievable sensitivity of PCR targeting cppB is 94%. 20 The use of the 16S–23S rRNA spacer region as target overcomes this limitation.
U. urealyticum was separated into U. parvum and U. urealyticum in 2002. 6 It has been reported that U. parvum is isolated more commonly than U. urealyticum from healthy carriers. 21 Others have reported a higher prevalence of U. urealyticum (or one of its subtypes) among men with urethritis than controls. 22 These findings suggest that U. urealyticum is the more virulent species, but the evidence is still limited and inconsistent. In our study, U. parvum was detected in 72% and U. urealyticum in 40% of specimens from women. These results are consistent with the high rates of carriage of ureaplasmas in women, especially those with multiple sexual partners, but neither species is implicated in lower genital tract disease in women. By contrast, there is good evidence that at least one of the Ureaplasma spp. can cause non-chlamydial NGU. In our study U. urealyticum was found significantly more frequently than U. parvum in symptomatic men. While this is consistent with a causative role in NGU, further investigation will be needed to compare the rates of detection between men with urethritis and asymptomatic controls.
All other species targeted in this study were found infrequently, even in this relatively high-risk group of patients. The study was not designed to confirm the roles of these organisms in lower genital tract infections, but to develop a tool for use in larger, controlled, studies that would be required to do so. Recently, a number of other mPCR-based methods to detect several genital pathogens simultaneously in a single assay have been reported. They include an mPCR which can detect N. gonorrhoeae, C. trachomatis, U. urealyticum and M. genitalium in the same reaction tube without loss of specificity. 23,24 Until recently, the number of targets that can be amplified and distinguished in a single reaction, without loss of sensitivity has been limited. We have demonstrated in many previous applications that up to 40 separate targets can be amplified and distinguished using mPCR/RLB. 12 This assay could be expanded to include additional known or putative genital pathogens, without loss of sensitivity. It is relatively inexpensive and easy to perform, does not require costly equipment and is a potentially useful tool for diagnosis and epidemiological investigations of sexually transmissible infections.