
Abstract
Analytical toxicology is a complex discipline. Simply detecting a poison in a biological sample does not necessarily mean that the individual from whom the sample was obtained had been poisoned. An analysis can prove exposure and perhaps give an indication of the magnitude of exposure, but the results have to be placed in proper context. Even if sampling was antemortem an analysis does not necessarily prove the effects that the drug or poison had on the victim immediately before or at the time of sampling. Tolerance is one big issue, the mechanism of exposure (how the drug got into the body) is another, and of course with postmortem work there are always additional considerations such as site of sample collection and the possibility of postmortem change in analyte concentration. There are also questions of quality and reliability, and whether a particular analysis and the interpretation placed upon the result are appropriate in a particular case.
It is my very great pleasure to introduce Professor Bob Flanagan, who is Consultant Clinical Scientist at the Toxicology Unit at King's College Hospital NHS Foundation Trust. His particular interests have been analytical methods, treatment of mental illness, especially as regards use of antipsychotics, and the diagnosis of substance abuse, and on googling him today I gather that he helped identify volatile substance abuse, ie glue sniffing, as an emerging problem in the UK in the early 1980s. He is a Council member of the British Academy of Forensic Sciences and he has acted as a Consultant to the United Nations Office on Drugs and Crime and to the World Health Organisation. So it is with great interest that I look forward, as I am sure we all do, to hearing from him about
I thought I would start off with something I am sure you will understand. To paraphrase Paracelsus (1493-1541), “the dose makes the poison”. Carl Linnaeus (1707-1778), the founder of taxonomy, refined the definition of a poison thus: “it is not only the nature of a substance, but also the dose which distinguishes foods from toxins and medicines from poisons”. That really is the nub of trying to diagnose poisoning - just detecting a poison doesn't mean that someone has been poisoned.
Alfred Swaine Taylor (1806-1880) proposed a further definition “a substance which when taken internally is capable of destroying life without acting mechanically on the system”. Here he was of course distinguishing between poisons such as alkaloids or heavy metals, which act as “biochemical” poisons, and caustic substances such as sodium hydroxide and corrosive acids, for example concentrated sulphuric acid, which are “energetic” poisons. Radioactive elements too, although of course unknown to Taylor, are also capable of acting mechanically (energetically) on the system in addition to any toxic properties possessed by the intact atom or molecule of which they are part.
Taylor was a pupil of Sir Astley Paston Cooper (1768-1841) at Guy's Hospital and then of Mathieu Joseph Bonaventure Orfila (1787-1853) in Paris. Orfila of course we look upon as the founder of our discipline of analytical and forensic toxicology. While in Paris, Taylor attended lectures given not only by Orfila, but also by the chemist Joseph Gay-Lussac (1778-1850) and by the guy whose name I can never pronounce (Baron Guillaume Dupuytren [1777-1835]), but who has a famous contracture named after him! (Laughter.) Taylor was in turn lecturer and later Professor of Medical Jurisprudence at Guy's from 1831 to 1877. 1 He is credited with bringing together clinical and analytical observations and placing them in a legal context, and his books, notably The Principles and Practice of Medical Jurisprudence (London: Churchill, 1865), stayed in print through various editions until relatively recently. 2 Poisoning was perforce an important aspect of Taylor's work. In 1831-1846, for example, poisoning had accounted for about 45% of all cases in which the law required medical evidence. 1
Taylor's principal pupil and successor was (Sir) Thomas Stevenson (1838-1908). Taylor had become unofficial analyst to the Home Office during his tenure at Guy's. Stevenson became the first Scientific Analyst, later Senior Scientific Analyst, to the Home Office (1872-1908) and as such was involved in investigating nearly all the most notorious poisoning cases for some 35 years. 3 One of his assistants was John Webster (1878-1927), who went to work with (Sir) William Willcox (1879-1941, a past President of your Society) at St Mary's. Webster himself became Home Office analyst (1919-1927) when Willcox retired from that post, the first chemist per se to be the official analyst as opposed to a an individual who was also qualified in medicine.
Both Orfila (the case of Madam Lafarge, where his identification of arsenic was eventually vindicated) and Taylor (the case of Thomas Smethurst (1859), where Taylor's misidentification of arsenic was strongly criticised by the Bristol-based toxicologist William Herapath (1796-1868)), were no strangers to controversy. Stevenson was seemingly an astute analyst and did not arouse such feelings. Perhaps in part due to his position with the Home Office and perhaps also because of improving communications and transport as industrialisation took hold towards the end of the 19th century, he seems to have been involved in case investigation. He didn't just get samples through the post or by courier, test for poisons, and then send the results back for someone else to comment on. He helped guide police in their investigations as well as preparing statements for the courts, as evidenced by the casebooks held today in the Gordon Museum, now under the auspices of King's College London ( Figure 1 - the suspicious death of Edwin Bartlett led to the famous trial of his wife Adelaide, the allegation being that she murdered him by giving him chloroform). This ability to consult immediately with experienced toxicologists when investigating potentially complex cases and an appreciation of the need to let them see the whole picture is I feel something that we have lost today.
Stevenson Casebook 1886. Case of Edwin T Bartlett (Gordon Museum). 9 Jan - Received at Home a telegram from Bodmer (?); then he came and delivered the HO order no X8963 for analysis. Wired Coroner (Hicks) to send articles on 11th…

It is worth remembering that in Stevenson's day many things were different from present day practice. There was no system of Home Office registered forensic pathologists until the 1890s, for example, although even today the great majority of postmortem examinations are carried out by clinical pathologists with little formal forensic training. Poisons availability was however very different. There were fewer drugs than are available today, but some of those that were used were extremely toxic and mixtures were common. Inorganic poisons such as sodium hydroxide were much more widely available than nowadays, and poisoning with many such compounds gave rise to distinct clinical features. Analytical methods, however, were rudimentary and depended on chemical tests rather than the chromatographic or spectroscopic methods that rule the roost nowadays. This being said, sensitive tests for certain compounds, arsenic and chloroform for example, were available by the early 1900s. Finally, knowledge of drug metabolism, drug interactions, and the treatment of poisoning was likewise rudimentary.
Poisoning by aconite
Stevenson was involved in investigating at least two “murderous medics” as I call them, somewhat tongue-in-cheek. Murderous medics (strictly murderous healthcare professionals) are nothing new4,5 One of Stevenson's cases was that of Dr Thomas Neill Cream (the “Lambeth Poisoner”), who in 1892 poisoned by strychnine Mathilda Clover in Lambeth Road. After three similar deaths of prostitutes 1891-1892, her body was exhumed and the true cause of death established. Stevenson also helped investigate the case of Dr George Lamson (the “Sleight of Hand Poisoner”). Lamson was a morphia addict (shades of Dr Harold Shipman, who was known to have abused pethidine early in his career). In 1882 Lamson was found guilty of murdering his 18-year-old handicapped brother-in-law Percy Malcolm John in 1881 by giving him aconite, which contains a mixture of highly toxic alkaloids including aconitine ( Figure 2 ), which occurs together with other alkaloids in various Aconitum species including Aconitum napellus (monkshood, wolfsbane).
Structural formulae of aconitine, jesaconitine, and pseudaconitine.

This was the last documented case of homicidal aconite poisoning in England until the “poisoned curry” incident, the murder of Lakhvinder “Lucky” Cheema and the poisoning of his new girlfriend Gurjeet Choongh, in Feltham, West London, in 2009. However, further research has unearthed reports of two cases from elsewhere. Mori et al 6 described a homicide in which the related alkaloid jesaconitine ( Figure 2 ; also found in A. napellus) was detected in the vomit, stomach contents, plasma, and urine of the victim at concentrations of 32.2, 5.48, 0.43 and 1.07 mg/L, respectively. The total amount of jesaconitine in the stomach contents was 1.3 mg. Secondly, Van Landeghem et al 7 described a case that was thought initially to be death by strangulation, and only discovered to be a homicidal poisoning some five years after the event. On further interrogation, a woman confessed to having murdered her husband by homogenising three Aconitum plants in red wine. Additional toxicological analyses using liquid chromatography-tandem mass spectrometry (LC-MS/MS) demonstrated the presence of aconitine in urine, liver, and kidney from the deceased at concentrations of 0.81 mg/L in urine, and 6.5 and 1.3 μg/kg in liver and in kidney, respectively.
The Feltham case came to trial in 2010. According to Miss Choongh, she and not-so-Lucky Cheema were sharing a meal and were talking of wedding plans (The Sun, 8 January 2010), when Mr Cheema, 39 (remember this is The Sun), said “I'm not feeling very well. My face has become numb; when I touch it I cannot feel it.” Miss Choongh (19) recalled having a shower and then beginning to feel similar ill effects, and Mr Cheema called an ambulance. Miss Choongh became comatose in hospital (I remember talking to one of the ITU team at the time and I was told she suffered every possible type of cardiac arrhythmia before they managed to stabilise her) and was ventilated for two days before recovering, but Mr Cheema died within two hours of admission. 8
Of course the medical team did not know what they were dealing with, and neither did they have the full history Miss Choongh gave when she recovered. However, the almost identical presentation of two hitherto well young people from the same household in such a serious condition clearly suggested a common aetiology, be it chemical, biological or microbiological. The main symptoms reported initially were copious and continued vomiting, visual disturbances, muscle weakness, dilated pupils, and unusual cardiac arrhythmia. Blood pressure and other observations were normal on presentation, but tachycardia and metabolic acidosis developed quickly. The patients did not seem disoriented and did not suffer convulsions.
At King's College Hospital we were contacted and asked to screen for toxic metals (principally arsenic) and aluminium phosphide as a matter of urgency. We looked for arsenic and mercury in the sure knowledge that there would be nothing of consequence to be found since the clinical features as reported to us, as well as the rapid onset of the illnesses, did not fit with acute exposure to toxic metal salts. Aluminium phosphide could of course not be tested for, but likewise seemed to be excluded by the clinical presentation.
In discussion with the medical team I suggested an urgent digoxin immunoassay to exclude oleander poisoning since the clinical features as described to me fitted with oleander at least in part, and there was no knowing whether a mixture of poisons had been ingested. Both common oleanader (Nerium oleander) and yellow oleander (Thevetia peruviana) contain car-diotoxic glycosides (neriifolin, thevetin A, thevetin B, and oleandrin) and poisoning with these plants or extracts of the plants is relatively common in India and in Sri Lanka and is often fatal. 9 It is well known not only that these glycosides cross-react to a greater or lesser extent on most if not all digoxin immunoassays, but also that anti-digoxin Fab antibody fragments, licensed for treating digoxin poisoning, are effective against a range of other glycosides including oleander glycosides. 10 However, I was told that nothing was detected by the assay thus excluding oleander as far as I was concerned, and I heard no more about the case for several months.
Clearly foul play was suspected at a very early stage, with concern focused on the curry that the partners had been eating immediately prior to admission to hospital, the remains of which were seized by police. Moreover, Mr Cheema had spent a week in hospital the month before having presented with headache, vomiting that had not responded to the usual antiemetics, and numbness of the hands and feet, a history that was available at the time of the second admission. He had been investigated for subarachnoid haemorrhage, but in the event no diagnosis was made. Postmortem findings were unremarkable with oedematous lungs and some corrosion of the oesophagus that could well have been due to continuous vomiting.
I was given to understand that the forensic toxicologists instructed by the police spent some time searching for oleandrin and other toxic glycosides in the remains of the meal, all to no avail as toxic glycosides seemingly had already been excluded as discussed above, but this information may not have been passed on. Later I was informed that apart from the alkaloids sanguinarine and chelerythrine, found together in Chelidonium majus (Greater Celandine), a plant that is not noted as being particularly toxic, nothing else had been identified on conventional drug screening procedures. Given that oleandrin, etc had been excluded, I had long been of the opinion that a toxic alkaloid was the only possible poison that could have caused the clinical features described so quickly after the presumed time of exposure, so in discreet consultation with Dr Randall Baselt I passed on a list of possible candidates that included aconitine, emetine, solanine, taxine and yohimbine. Dr Baselt particularly suspected emetine, a constituent of syrup of ipecacuanha (ipecac), since he remembered two case of deliberate child poisoning that he had investigated that involved this drug.
Again some months passed until I was again contacted with the news that aconitine-type compounds had indeed been found in the curry using LC-MS/MS and further work was being undertaken to identify them. Comparison with aconitine pure compound showed that although these substances appeared to be related to aconitine, they did not match with aconitine itself or with the expected hydrolysis products of aconitine. Subsequently, a comparison of MS data with data in a paper describing the detection of yunaconitine in urine from suspected aconite poisoning cases revealed the presence of not only yunaconite, but also pseudaconitine, indaco-nitine, bikhaconitine, and deacetylyunaconitine. At this time too it seems that the information from Miss Choongh as to the symptoms that included para-esthesia she and Mr Cheema experienced before attending hospital was made available, symptoms similar to those Mr Cheema had experienced on his first admission, adding to the likelihood that an aconitine-type compound was involved.
In the meantime I had recommended Professor Robin Ferner to comment on the clinical features that might be expected in aconite poisoning, and it was he who suggested that one of the compounds in question might be pseudaconitine ( Figure 2 ), which is found together with other alkaloids in Indian aconite (A. ferox), for example. 12 This indeed proved to be the case, and pseudaconitine and related compounds were found in the remains of the curry, in blood from Mr Cheema, and in plant fragments from the suspect's handbag. In all 17 Aconitum alkaloids were detected in the remains of the curry Mr Cheema and his fiancée had been eating. 8
The oral lethal dose of aconitine itself is thought to be of the order 1.5-6 mg by mouth. Pseudaconitine is thought to be even more potent. Aconitines are quickly absorbed if given orally. Paraesthesiae, including tingling in the mouth, soon extend from the extremities to the whole body. Loss of sensation, sweating, nausea, vomiting, severe pain, muscle cramps, and diarrhoea may ensue. Hypotension, bradycardia, cardiac arrhythmias, and respiratory paralysis follow in severe cases.13–16 Treatment is normally symptomatic and supportive, and may include repeat-dose oral activated charcoal. Use of magnesium sulphate has been described. 17 Aconites are common constituents of Chinese herbal medicines. 18 There have been a number of reports of fatal poisoning (accidents or deliberate self-poisoning) with A. napellus. Indeed, Dr George Edward Male (1779-1845), looked on as the father of English medical jurisprudence, is thought to have died as a result of taking tincture of aconite for pain relief. 19
The concentrations of aconitine reported in postmortem femoral blood and urine were 10.8 and 264 μg/L, respectively, in one case. 20 The aconitine concentrations in antemortem urine and serum were 334 and 6 (estimated) μg/L, respectively. Pullela et al 21 using LC-MS/MS reported aconitine concentrations of 3.6 and 149 μg/L in postmortem blood and in urine, respectively.
Unfortunately no quantitative data are available on the Feltham case simply because no reference sample of pure pseudaconitine was available with which to perform instrument calibration. This is a problem with other rarely encountered poisons and also with newly emerging “designer” drugs, which have become an especial concern in recent years. One suggested solution is to use liquid chromatography with chemiluminescence nitrogen detection,22,23 which enables calibration on the number of nitrogen atoms in a molecule, but this is not ideal since the instrument response may differ at different times in the chromatogram due to the presence of co-eluting compounds, for example.
These considerations notwithstanding, the Feltham case does emphasise the limits of modern instrumentation, powerful though it is, and shows that there is still a need for experienced analysts/toxicologists to guide use of the instruments! As to the outcome, Mr Cheema's ex-girlfriend Lakhvir Singh was convicted of his murder and causing grievous bodily harm with intent to Ms Choongh, as reported in The Sun. Last December I was contacted by police who suspected that aconite may have been used in another murder in West London. I referred them to the analyst in the Feltham case, but apparently no analytical evidence was forthcoming to substantiate their suspicions.
A chloroform-related death
Early one morning in February 1999, a 34-year-old lady was found dead, fully clothed, in a garden close to her house in Broxbourne, near Livingston, West Lothian. She had recently learned that her 30-year-old partner had got another woman pregnant. She herself was a pub manager and a known amfetamine user, but was not thought to drink alcohol and was not known to abuse other drugs. The police were of course involved and a pathologist was called to the scene, but nothing was adjudged especially suspicious.
The postmortem examination revealed trivial external injuries, but no evidence of natural disease that could account for death. The lesser curve of the stomach showed evidence of acute haemorrhagic gastritis, with no ulcers or erosions. No unusual smell was noted on opening the stomach. Thorough microscopic examination of the main organs of the body, including liver and kidney, and the brain disclosed no significant pathological or histological changes, although occasional contraction band necrosis was noted in muscle from the right ventricle. The cause of death was given as hypothermia secondary to amfetamine intoxication because a very small quantity of amfetamine (0.05 mg/L) was found in postmortem blood. Cannabinoids were also detected. No injection marks were identified on the body, which is an important point we will come back to later. It is of note that as I understand things the toxicology laboratory was told only to look for common drugs of abuse and alcohol, not to look for poisons per se. If there had been a broader search for poisons then the subsequent course of events is likely to have been very different.
The deceased was found lying on her back with traces of vomit round her nose and mouth. The defence were told that it went to cold case review with Dr Iain West (1944-2001), who is said to have commented when looking at photographs of the scene, “Oh, that's suspicious, lying on her back like that”, or words to that effect. I don't know about such things as I have never investigated a crime scene. But looking at the photographs you can see that round her nose and mouth are obvious marks of vomit. So the vomit had been cleared and it wasn't at the scene, and yet the death still wasn't looked upon as suspicious, it was just signed off and consigned to history.
Well now, some questions even at this stage were: Why so close to the house? It was only just down the common path; there is a house either side and there is a shared path down the middle. Why on her back? And she had got her knickers on back to front. Some girls say, “So…?” Some say, “Oh, that's suspicious.” I don't know. I'll leave that to a vote among the ladies later. Why no vomit at the scene? That was an important thing to me. But there were traces of vomit in her nose and behind her front teeth. And the cold case review, we were told, said there was a burn, possibly a chemical burn, on her lip. From looking at the photographs it is not clear to me that the “chemical burn” on her lip was not just a cold sore. Similarly, to me there is absolutely no indication that there are marks indicative of a pad being placed over her nose and mouth, which is what we were told Iain West said he could see.
The story that came out was that a “Crimestoppers” call reported that her partner, who was a self-admitted cannabis user, is said to have confessed the day after the death to murdering her with chloroform and insulin, and also is said to have made this confession to a second friend. The conversations were only reported to police about 17 months later, which I think is suspicious straightaway. You inevitably wonder what has been going on in the meantime. Anyway, by pure chance it transpired that the forensic laboratory had not cleared out their refrigerator and freezer, so the samples were still there 18 months later. Remember, this was a case that had been confined to history. So they reanalysed the postmortem blood and vitreous humor, and reported a blood chloroform concentration of 32 mg/L. Note that insulin was mentioned as well as chloroform. Now, obviously insulin can't easily be tested for postmortem, especially 18 months after the event. Moreover, no injection marks were noted at the postmortem examination, although this does not preclude injection with a fine needle. There was also the question of where the alleged perpetrator could have obtained these compounds and also how he would have known how to administer them.
Anyway, based on this new information the partner of the deceased was arrested and charged with murder. The indictment libelled specifically that he had rendered the deceased unconscious by forcing her to inhale chloroform and then placed her outside where she froze to death. Under Scots law a criminal charge cannot be proved without corroboration. This means that there must be evidence from two or more sources pointing to the accused as the perpetrator of the crime. In this case the main evidence related to the confession made by the accused to his two associates. It follows that the accused presented the source of the evidence and although the confession was allegedly made to two persons there was only one source, namely the accused. However, where a confession made by an accused person contains details of a crime that could only be known by its perpetrator then these details (referred to in law as “special knowledge”) provide the necessary corroboration of the essential facts that a crime has been committed and that the accused is the perpetrator. This placed an obligation on the prosecution to prove that chloroform had entered the body of the deceased by the method disclosed in the alleged confession.
Well, this came to me, obviously, two weeks before the trial. I say obviously - you know lawyers… How many lawyers in the audience? (Laughter.) It came to me via the defence two weeks before the trial was due to commence. I was initially very suspicious of the analytical evidence firstly because homicidal chloroform poisoning these days is very rare. Why is it very rare? It went out of fashion really a hundred years ago, because a simple test for chloroform became available. And nowadays chloroform is not easily available for “end-user applications” because it is a potent ozone-depleter among other things. But of course the laboratory didn't do the simple test because as I have indicated I suspect that they were not told to look for poisons per se, just to look for common drugs of abuse and alcohol. Secondly, 32 mg/L is not necessarily fatal. In anaesthesia you can get up to 200 mg/L and, under proper conditions, the patient recovers quite happily, although the risk of something going wrong is much greater if the vapour is inhaled either voluntarily under uncontrolled conditions, or under duress, as I well know from my work on volatile substance abuse.
Further factors with chloroform are that it is ubiquitous in the environment. I could measure chloroform in a blood sample from any of you given appropriate equipment, at anything up to 5 μg/L, possibly slightly higher, because chloroform is in our water supply in very small quantity. It comes from use of chlorine at the water treatment works, and there are lots of other sources as well. You can also get chloroform formed on headspace gas chromatography (50°C or so) as an artefact from trichloroacetate, which is a metabolite of some drugs, notably chloral hydrate, and is also a metabolite of another solvent, trichloroethylene. Finally, the chromatograms from the analysis of the vitreous humour suggested that 1,1,1-trichloroethane, which is another solvent that can be metabolised in small amount to trichloroacetate, might be present.
Interestingly, in the Scottish system the defence and the prosecution have equal access to each other's witnesses before the trial (“precognition”), so to my simple mind it is adversarial, but investigational at the same time. So I went up to see Her Majesty's Advocate Depute (prosecuting counsel) soon after the trial had started, but before I had given evidence, and said exactly what I had said to the defence team. I thought this was great because I was obviously acting for both sides equally, or so I thought, which is just what an expert witness should do. Be this as it may, I suggested that stomach contents and liver from the deceased, which we were told had been stored at — 20°C since their arrival in the laboratory some two years previously, should be analysed to confirm the blood results. The liver sample, although we did not know from whereabouts in the liver it had been obtained (liver for toxicological analysis should ideally be obtained from the right lobe as this is furthest from the stomach), I suggested should be given priority. I said this in essence because (i) the blood may have lost chloroform since for one thing it was likely to have been opened during the course of the original analysis (chloroform is of course volatile), (ii) there is always the risk of cross-contamination of liquid samples especially with volatile laboratory solvents (chloroform was commonly used in laboratories at that time), and (iii) loss of chloroform and cross-contamination are much less likely with solid samples, especially if stored frozen in a sealed container.
Her Majesty's Advocate Depute did arrange for the analysis of the liver (but not the stomach contents) and the result was given to me on the morning I was due to give evidence. The result as stated was 1064 mg/kg, which is an enormous quantity, about 0.1% of liver weight, far higher than any other chloroform poisoning deaths reported in the literature. In retrospect I should have heard alarm bells ringing, but at the time my reaction was that to achieve such a high concentration in liver simply by inhalation was just not possible, ergo she must have ingested a fair proportion, if not all, of the dose. It is well known that chloroform is much less toxic by ingestion than by inhalation, liver and other tissues absorbing some chloroform before any drug gets to the brain. Nevertheless the presence of such a vast quantity of drug in the liver clearly pointed to chloroform poisoning as the cause of death. Ingestion would, of course, be a possible explanation for the gastritis found postmortem, since liquid chloroform is an irritant poison and can cause reddening of mucous membranes, for example. Of course this slant on things completely undermined the prosecution libel (in Scots law a libel is a formal statement of a charge) that exposure had been totally by inhalation.
Again, interestingly, in Scottish law as an expert witness you are not allowed to sit in and hear the evidence of other experts, even those of your own side, unless by express permission of the court. It is thus not like in England, where having the expert(s) for the other side in the court can sometimes help guide cross-examination and in other areas. In Scotland it doesn't work like that and you are treated as a witness as to fact. I think the English system is better in this respect, although as I have indicated I think the Scottish precognition system has advantages.
So, in my absence, the prosecuting counsel said, “Oh, there were clear marks on the face showing that a chloroform pad had been put to the mouth”, or words to that effect. The pathologist who had performed the original postmortem had by this time totally been converted to the chloroform-by-inhalation scenario and asserted that chloroform is a corrosive poison, hence there would have been massive corrosion of the gastrointestinal tract if the deceased had ingested chloroform, and perforce this would have been obvious at postmortem. That is clearly wrong of course because chloroform is not a corrosive poison, as noted above. I had liquid chloroform in my mouth several times many years ago - mouth pipetting, not allowed nowadays! It stung a bit, had a mildly sweet taste, but didn't do any damage as far as I'm aware. Even more amazingly, the pathologist claimed that the liver chloroform concentration must be so high because it had concentrated chloroform out of blood after death. This was clearly wrong - after death as membranes, etc break down poisons tend to diffuse from areas of high concentration to areas of low concentration, but not the other way about. The only way the liver concentration could have increased after death was if there had been a large amount of chloroform in the stomach or intestines at death, which was clearly something the Crown disallowed. Flawed evidence on two counts, but placed before the jury nevertheless.
In the event the accused was convicted, but by a majority verdict. In Scotland there are 15 jurors and they accept a majority, so eight to seven would have achieved conviction. Clearly the defence had ample grounds for appeal, but this took seven years to be heard! In the event the Crown admitted, after intervention from Professor Bob Forrest, that they had over-stated the chloroform concentration in the liver at trial 1000-fold. The true concentration was actually 1 mg/kg, not 1000 mg/kg. No explanation was ever forthcoming for this error. Of course 1 mg/kg is extremely low, and does not suggest chloroform exposure from sources other than drinking water, for example. It later transpired that the liver had been allowed to thaw completely and the container had been opened when it was sent to another laboratory for pesticide analysis (why this was done was never made clear) so its evidential value had been nullified even before the original trial.
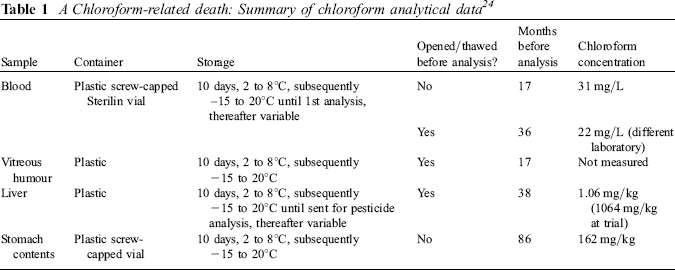
The Crown, by this time, had agreed to the analysis of the stomach contents, which we were assured had been stored frozen since receipt in the laboratory at the time of the original investigation. The result was 162 mg/kg, which is far higher than the blood concentration as reported. This time the evidence was that ingestion was a likely route of at least some, if not all, of the exposure. A summary of the analytical data is given in Table 1.
A Chloroform-related death: Summary of chloroform analytical data 24
There was of course other evidence. It seemed clear that the body may have been dressed and moved after death. The cleared vomit was never found. There were no signs of liver damage at postmortem. Why is that important? Well, if she had been abusing chloroform chronically, then I would expect there to be overt liver damage because that is documented in people who have died after periods of chloroform abuse. 25 Chloroform is very toxic to the liver. There was also evidence of urine under a new carpet in the living room of the accused's house, the room where the murder was said to have been committed, but that could not be traced back to the deceased. There were also the reported confessions.
Finally, when it came to the appeal hearing in front of three Law Lords, this again was interesting because when I gave evidence Her Majesty's Advocate Depute (not the original prosecuting counsel, they are three-year rotating appointments), while trying to cross-examine me, was actually instead cross-examined rather sternly by their Lordships. In the event, after a five-day hearing, the conviction was quashed and the Crown was refused permission to bring a further prosecution.
What really had happened? Was she fatally poisoned? She was obviously exposed to chloroform, but was it enough to kill her? Did insulin pay any part? I am a bit suspicious about the role of chloroform. One thing that didn't really get discussed was where the chloroform came from. Would anyone like to hazard a guess where the chloroform came from that was used, assuming it was used, and who if anyone supplied it?
A
All this of course has much on common with the suspicious death of Edward Bartlett and the subsequent trial of his wife Adelaide for murder in which Sir Thomas Stevenson was a key player, as noted above. Ironically perhaps in view of the Broxbourne case, Adelaide was charged with administering liquid chloroform to Edward. An article by Stevenson giving his own view of the case obtained via Professor Keith Simpson (1907-1985) was published posthumously in The Criminologist. 26 Important evidence was the presence of a large amount of chloroform in the stomach at postmortem and the marked inflammation of the stomach lining. In conclusion, it is perhaps appropriate to recall the famous remark of Sir James Paget (1814-1899) of St Bartholomew's Hospital made after Adelaide's acquittal: “She should tell us, in the interest of science, how she did it.” 27
Fire retardants and sudden infant death syndrome
So we have had one clear poisoning (two victims) and one probable poisoning, although the circumstances of the latter case and indeed the exact cause of death remain unclear.
Do any of you remember the fire retardants and sudden infant death syndrome (SIDS) scandal? In 1989 a chemist, I'm embarrassed to say, vouchsafed that he had research data that showed that toxic gases (arsine, phosphine, and stibine) released by fungal metabolism of fire retardants in cot mattresses were the cause of many of these deaths.28,29 The fungus was supposed to grow on saliva from the babies themselves, a theory first propounded in New Zealand in the mid-1980s by a Dr Jim Sprott. It doesn't make sense, does it, if you think about it without knowing any of the background? I know from my own investigations into volatile substance abuse that you have to try very hard to achieve intoxication, let alone kill yourself, by deliberately inhaling pure gas or concentrated vapour, let alone be accidentally fatally poisoned in an open environment by minute quantities of albeit toxic as opposed to asphyxiant gas slowly released in the way suggested. Shades of Stevenson again of course as arsenic (for example the Maybrick case of 1889) and antimony (as potassium antimony tartrate, Tartar emetic, case of George Chapman, the “Borough Poisoner”, 1903) were analytes he was not unfamiliar with, although exposure was usually, if not invariably, by ingestion in the cases he dealt with.
Anyway, as you may remember there was an enormous fuss over the toxic gas hypothesis. It cost the Department of Health many thousands, if not hundreds of thousands, of pounds in trying to set up objective studies to test the hypothesis and caused immense distress to thousands of parents in this country and indeed in other parts of the world. On my recommendation a colleague from Holland and expert on metals toxicology wrote an editorial for the British Medical Journal that reviewed the evidence, such as it was, and he came to the conclusion that there was in fact no evidence at all. 30
Unfortunately, the Cook Report got in on the act and looked to generate evidence to support the hypothesis, and in the event cooked the books …
The Lancet published a letter wherein it was claimed that mean antimony concentrations in liver samples from SIDS cases (sudden unexpected death in infancy, SUDI, to give it its current name) were very much higher than in “controls”, i.e. samples from children of equivalent age who died from causes other than SIDS. The work was, I suspect, funded by the Cook Report. The theory went that the antimony was derived from stibine (SbH3) inhaled by the SIDS victims before death. They didn't look for arsenic, because arsenic is in all of us, mostly as non-toxic species, and they didn't look for phosphorus because likewise phosphorus is ubiquitous in living systems as phosphates. Antimony, however, is nowhere near as prevalent and so they looked for antimony. I am confident the analyses were reliable because they were performed by a well-respected analyst. It was the interpretation placed on the results that was fatally flawed. The report cited data from liver samples from 30 SIDS cases 1986-1988 and seven from 1993 to 1994. The mean liver antimony concentration (N =20) was reported as 7.1 μg/kg. Micrograms per kg in ratio terms, parts per …
A
What I did, which I don't think anybody else did, was to talk to the author and find that results from paired blood samples were also available. The mean blood concentration in 19 SIDS cases was 2 μg/L (a litre of whole blood weighing 1.06 kg), while antimony was only measurable in two blood samples from different “controls” at concentrations of 0.60 and 0.90 μg/L, respectively. I further reasoned that if the blood and liver data from the SIDS cases really reflected antimony exposure in life then there should be a correlation between the individual results. The plot of the original data, kindly supplied by the author, is shown in Figure 3 . Not only is there no correlation, but also it can be seen that the data are skewed dramatically. In one sample both liver and blood values were high, one had a high liver and low blood, and one had a high blood and low liver, with the rest of the results clustering down near the limit of measurement. The means as reported thus grossly misrepresented the data as a whole - so much for The Lancet, statistical advisers, etc. If the outliers are excluded the results are similar to those seen in non-SIDS deaths.32,33 Given that the samples had not been collected with the analysis in mind, sample contamination was the obvious likely explanation for the outliers even in 1996.
Plot of blood and liver antimony data from SIDS cases
31
(R = 0.14, N= 35).

Did anybody ever meet Roger Cook, by the way? I always used to think he must be quite a nice bloke when I heard him on the radio exposing this, that and the other fraud. In reality when I actually met him he was very well built (we couldn't actually get him to fit in a lab coat) and I started to feel somewhat sorry for his victims. Anyway, in responding to the 1998 Report of the Expert Group chaired by Lady Limerick set up to look into the “Toxic gas hypothesis”, an investigation that lasted three-and-a-half years and concluded that the theory was in essence simply rubbish, Joyce Epstein, Secretary-General of the Foundation for the Study of Infant Deaths, commented thus (http://sids-Network.org/experts/toxic_gas_hypothesis.htm, accessed 20 August 2012):
“The toxic gas theory has now received the most thorough possible attention, and has been rejected as unfounded. It brings to a close a ghastly episode in public health scare-mongering prompted by The Cook Report in 1994, which broadcast its programme without responsibly substantiating the evidence.
Thousands of parents were distressed and misled by the story, convinced that mattresses kill babies. Even now a survey shows that more parents think mattresses cause cot death than are aware, for example, that smoking in pregnancy is dangerous for babies.”
Could this sort of thing happen now?
A
This gross misrepresentation of analytical data happens in other areas of course. A common ploy is to send samples (blood or urine) to an “environmental toxicology” laboratory and provide interpretation based on “population averages” or some other meaningless figure, or do a dimercaptosuccinic acid (DMSA) challenge test to demonstrate enhanced urinary excretion of “toxic metals”, a test that of course also risks depleting the body of essential trace elements. As an example, Table 2 gives real values for a “toxic solvents screen” reported a few years ago on an individual who had been convinced that she was being poisoned by her environment. I could give examples where blood concentrations of organochlorine pesticides are similarly misrepresented.
Results of a “solvent screen” on a patient blood sample sent to an environmental toxicology laboratory
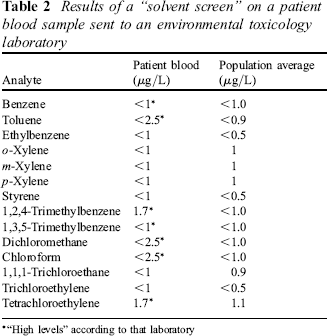
“High levels” according to that laboratory
You can see the “population average” column. The units are μg/L, i.e. parts per thousand million. There are figures there, but no indication of the demographics, occupation, geographical location, etc of the population studied, and no indication of the range of values encountered in that population. Moreover, how you can get an average to be <1, for example, escapes me. The truth of the matter is that these measurements are at the limit of sensitivity of the assay procedure - you can see that by comparing the two columns with < 1 being reported in one column with a population average of 1.0 in the next column, for example. But a few measurements are starred to denote “high levels” and patients often take this as confirming their belief that they are being poisoned by the highlighted agent. In fact, nothing could be further from the truth - given appropriate instrumentation I would be able to measure similar concentrations in all of you, possibly slightly higher. We have already seen that “background” blood chloroform concentrations in this country can range up to 5 μg/L quite easily, yet in this report “<2.5” is said to be “high”.
There is a US organisation called Quackwatch that aims to combat health-related frauds, myths, fads, fallacies, and misconduct with focus on providing quackery-related information. Its website www.quackwatch.org (accessed 20 August 2012) gives information on unproven or ineffective remedies/ treatments. The site contains articles and other information criticising many forms of alternative medicine and indeed “alternative laboratory medicine”. Similarly, HealthWatch (www.healthwatch.org.uk, accessed 30 August 2012) in the UK assesses the effectiveness - or not - of complementary/alternative and conventional treatments.
Mental health and poisoning
The incidents I have discussed thus far have perforce been historical in nature, although I trust you have seen that there are lessons to be learnt from how they were handled. The interface if I may call it that between mental health and poisoning has always been one that coroners, for example, have encountered frequently. Leaving aside the question of substance abuse, there are at least three factors at play. Firstly, there is the risk of self-poisoning, and if the patient dies the coroner may have to pronounce upon intent. Secondly there is the question of drug treatment, the cornerstone of the treatment of mental illness nowadays. Modern antidepressants and antipsychotic drugs are potent and often effective if used appropriately, but there is always a risk of unexpected effects such as the increased risk of suicidality said to be apparent with paroxetine in major depressive disorder. 34 There is also a risk of side-effects from most such drugs in normal use, one of which may be prolongation of the QT interval hence the risk of precipitating a fatal arrhythmia. A fatal arrhythmia leaves no evidence postmortem of course, and can occur in the absence of any drug treatment. Effective as they are in many cases if used appropriately, overdose with these drugs, be it accidental, deliberate, or iatrogenic, can prove fatal. Finally, patients with serious mental illness, treatment-resistant schizophrenia for example, are thought to have a higher risk of sudden death than people in the general population irrespective of treatment. 35 Whether this is related to the fact that some 85% of people with schizophrenia smoke if given the chance I do not know.
New drugs are of course viewed with suspicion as regards possible toxic effects, which is sensible in a way, but sometimes obscures the picture. With sudden, unexpected death in epilepsy (SUDEP), for example, new antiepileptic drugs were viewed with suspicion for many years until it was pointed out that the death rate before the new drugs came along was just the same as it was afterwards. Manufacturers, of course, are usually unhelpful, because they don't like it said that their drugs might kill somebody, and if incidents are reported to them that triggers the generation of massive amounts of paper that disappear off into the black hole that is the Medicines and Healthcare products Regulatory Agency (MHRA). I myself always look at the overdose toxicology of a particular drug before pronouncing on a particular set of circumstances leading to a fatal outcome in which the drug has been implicated. If it does not seem to kill when people deliberately take it in overdose, it seems to me less likely to kill in less straightforward circumstances.
With antipsychotics, concerns as to cardiotoxicity in normal use of thioridazine led to its voluntary withdrawal from general use in 2005, while older, “typical” drugs such has chlorpromazine and halo-peridol have gradually been superseded by more modern (and more expensive while on patent) “atypical” or second generation drugs. As far as I can see all the fuss about thioridazine generated by the Newcastle group36,37 has had no impact on the frequency with which antipsychotics are mentioned in connection with deaths notified to the coroner: other drugs are simply mentioned in its place. 35 In a nutshell there is and indeed was at the time no evidence that thioridazine caused more deaths than other antipsychotics. A classic mistake of not looking at the whole picture, and a very expensive mistake to boot. In the year after an urgent cascade fax from the Committee for Safety of Medicines (December 2000) warning of the dangers of thioridazine following the first Newcastle report (see ref. 36 - The Lancet again), the cost of primary care prescribing of atypical antipsychotics in England increased by £32 million. Over the same period, the prescribing cost of typical antipsychotics (included thioridazine) fell by just under £900,000. 38
And then there is the question of adherence to treatment. With many antidepressants, for example, there is an increased risk of withdrawal effects, which may include an increased risk of suicidality. 39 Secondly, many centrally acting drugs are much more toxic in overdose if a patient has not acquired tolerance to at least some of the effects of the drug through recent exposure, and of course there is the risk that the illness will reassert itself if treatment is not adhered to. On the other hand, if a prescribed drug is not found on analysis and the patient has clearly committed suicide, for example, or murdered someone, then this brings into question the quality of care they had received prior to death or other event.
The problem of assessing adherence brings me back to the thorny question of postmortem toxicology. It used to be thought that the concentrations of drugs measured in samples of blood, for example, reflected the situation perimortem. Thanks to the work of Professor Derrick Pounder in Dundee and others, we now know that this is anything but the situation for many drugs and other poisons.40,41 Most drugs used to treat mental illness have a large volume of distribution, ie during life on chronic dosage they are present in most tissues at far higher concentrations than in blood. After death as autolysis proceeds and membranes break down, blood concentrations of many centrally acting drugs tend to increase, a phenomenon perhaps more noticeable in blood from central sites such as the heart or vena cava than in peripheral sites such as the femoral vein.
Thus if a patient has been adherent to therapy measuring a somewhat higher concentration of many drugs in a postmortem sample, even a femoral sample, than might be expected for a given dose does not necessarily mean that an overdose had been ingested before death. For clozapine, for example, I expect on 4-500% increase in both clozapine and N-desmethylclozapine (norclozapine) concentration after death even with a femoral sample when the drug has been given chronically, although there is much variation.42,43 Olson et al 44 have reported similar findings with another centrally acting drug, fentanyl, in femoral blood.
Clozapine of course is always given orally while fentanyl may be given orally or transdermally. If the decreased had received a bolus injection of a drug such as risperidone in the gluteal muscle before death then this is not that far away from the femoral vein. Urine too may be a source of drug either for postmortem diffusion or sample contamination. Even if a relatively small proportion of a dose is excreted unchanged in urine, the urinary concentrations during life may be many times higher than the blood concentration.
In summary, all the available evidence must be taken into account when investigating a death if poisoning is suspected. An overall knowledge of the circumstances, time course, clinical/postmortem observations, poisons thought to be involved and their toxicology and metabolism, is important, as well as knowledge of the samples available for analysis and the analytical methods used. In my experience, perhaps because of the need to get on with postmortem examinations, sometimes not all relevant information is passed through to the laboratory tasked with the toxicological analysis to guide the priorities for the analysis and to inform interpretation of the results. This may cause serious problems later, as evidenced by the chloroform-related death discussed above.
An amisulpride-related death
To conclude with a final case example that brings together much of what we have discussed in the latter part of this talk. A 52-year-old man collapsed suddenly and died (2009). His only recent acute illness was an episode of vomiting and diarrhoea two days prior to death. He was prescribed lithium carbonate (1 g daily), the antipsychotic amisulpride (400 mg daily), and amitriptyline hydrochloride, an antidepressant (150 mg daily), all doses in accord with British National Formulary guidelines. He had been taking these drugs for some years and dosages were not changed in the months before death. At postmortem there were no tablet residues visible in the stomach, but the deceased was recorded as markedly obese with an enlarged heart (560 g) and an enlarged and fatty liver, which the pathologist in turn recorded as the immediate cause of death. However, postmortem blood was sent for toxicological analysis. Amitriptyline was reported as 0.28 mg/L, which is not far removed from what might be expected in plasma in life (nortriptyline was detected, but not measured). Lithium was 0.27 mmol/L, a bit low for plasma during successful treatment in life, but otherwise unremarkable, and amisulpride was reported as 13 mg/L. No tests were performed on stomach contents.
Plasma amisulpride might be expected to be 0.2-0.5 mg/L during chronic therapy, although concentrations up to 1.9 mg/L have been reported. 45 Even in volunteers given 400 mg by mouth we have likewise measured plasma amisulpride concentrations up to 1.9 mg/L four hours post-dose without ill effect. Anyway, the pathologist changed the cause of death to amisulpride toxicity simply on the basis of this one relatively high result in postmortem blood. The deceased's family, of course, said: “How on earth can this be? Why is the cause of death now poisoning when it was fatty liver? What has been going on? Does the fatty liver mean that the amisulpride wasn't being metabolised properly and the amisulpride had gradually built up and killed him? If this was the case, surely the clinicians treating him should have known about his liver condition and they should have cut the dose accordingly?”
The Procurator Fiscal was needless to say very concerned at this turn of events and sent the case to me for review. In response I pointed out that amisulpride is not metabolised; it is almost entirely excreted unchanged and thus its elimination from the body is not dependent on hepatic metabolism at all. Amisulpride and sulpiride are the only two antipsychotics this applies to. By the same token of course, if amisulpride had accumulated during treatment, why hadn't the same thing happened with amitriptyline?
Could the amisulpride result could simply be postmortem change? The problem here again was that the amitriptyline wasn't raised. Lithium distributes through body water and blood lithium concentrations are not thought to change after death except that after acute overdosage there may be some diffusion from the stomach or intestines, so no help on that score.
Amisulpride has been in clinical use since 1986 and is a very safe drug in normal use as antipsychotics go. Deaths after amisulpride self-poisoning are very rare. Postmortem blood amisulpride concentrations of 42 and 140 mg/L, respectively, have been reported in two deaths.46,47 In contrast, there are number of reports of survival after amisulpride self-poisoning with plasma concentrations in the range 7-23 mg/L, reliable values as sampled during life. 48 One patient survived the ingestion of an estimated 120 g amisulpride, some 100 times the maximum recommended daily dose of 1.2 g per day. 49 There are also two reports of sudden unexpected death in patients taking amisulpride,48,50 but I have long been suspicious about these. The only indications of amisulpride poisoning were the concentrations of amisulpride measured in postmortem blood (13.4 and 48 mg/L, respectively), and I wonder if these were simply the result of postmortem change or sample contamination, for example with urine; urinary amisulpride concentrations can be very high - we have measured up to 1380 mg/L in therapy.
Some years ago I reviewed another sudden, unexpected death in a patient treated with amisulpride. The deceased was a female patient in her 30s prescribed oral amisulpride and clomipramine (normal doses). She was looked after at home by her elderly parents and was found with no indication of overdose, self-poisoning, or anything suggestive of a cause of death. There was no history of recent illness such as could have caused renal impairment and the prescription of psychotropic medication had been stable for some time. The (femoral) postmortem blood amisulpride concentration was reported as 347 mg/L, with clomipramine and N-desmethylclomipramine (norclo-mipramine) 0.5 and 5.9 mg/L, respectively.
The pathologist recorded the cause of death as fatal amisulpride poisoning (“1000 times therapeutic”), the patient's general practitioner wrote to the MHRA labelling amisulpride as “dangerous”, and the case was referred by HM Coroner to the Serious Crime Unit (SCU) of the local constabulary for further investigation. The investigations of the SCU came to nought, but the parents were left not only devastated by the sudden loss of their daughter, but also shocked at the ensuing ordeal to which they were subjected.
This time, inspection of the laboratory records showed the measurements to be reliable as far as I could see. However, further information was available in the form of liver measurements, information that had been ignored by the (Home Office registered) pathologist. The liver (site of collection in the body not given) clomipramine and norclomipra-mine concentrations were 1.44 and 25.4mg/kg, respectively (290% and 430% higher than in blood, respectively). In contrast, the liver amisulpride concentration was 2.8 mg/kg (<1% of the blood concentration). The volumes of distribution (a measure of the likely distribution of the drug between tissues and blood during life) of both amisulpride and clomipramine are very similar. 51
In my opinion contamination of the blood sample, possibly from the bladder as discussed above, was the most likely origin of the obvious discrepancy between the blood and liver amisulpride concentrations. Only 300 μg (1/2667 of the daily prescribed amisulpride dose) added to 1 mL of blood would have been needed to give a blood amisulpride concentration of 300 mg/L. Further factors were that (i) there were no amisulpride tablet residues visible in the stomach at postmortem, (ii) if poisoning had occurred by ingestion then blood from the intestines would have travelled first to the liver hence higher liver concentrations than blood concentrations might be expected in such circumstances, (iii) there were no injection marks observed on the body at postmortem, (iv) injectable amisulpride was not available in the household, (v) amisulpride administration was supervised by the deceased's father, and finally (vi) there was no clinical evidence of renal or indeed liver dysfunction.
So in the end, I reported that sample contamination was the most likely explanation for the very high amisulpride result, a conclusion accepted by all concerned including the pathologist and the coroner. Of course the reason she died remained unclear, although it was possible that the combination of antipsychotic and tricyclic antidepressant together precipitated a fatal arrhythmia. But then again patients with schizophrenia have an increased risk of sudden death, as discussed above.
Anyway, to return to the gentleman I described earlier whose death was altered from fatty change of the liver to amisulpride intoxication, I said in summary: “I don't believe the cause of death can be stated as amisulpride toxicity solely on the basis of the postmortem blood amisulpride result, simply because amisulpride is a very safe drug in normal use and there is no clinical or other evidence of overdo-sage/poisoning.” In the event, the laboratory checked their figures and found that the postmortem blood amisulpride concentration should have been reported as 0.65 and not 13 mg/L (a 20-fold error in a calculation somewhere), so the cause of death was changed back to “fatty liver” and you would have thought everyone would be happy/relieved that the mystery had been solved. Not so unfortunately, because the family then wanted to know why the laboratory had made the error in the first place and in turn why the pathologist had changed the cause of death to amisulpride toxicity.
Now, normally I have much sympathy with pathologists when they are put in the position of having to assign a cause of death when the cause of death is perhaps not that clear-cut, although there is no evidence of death being due to anything other than “natural causes”. Relatives often expect answers even when there is no clear answer, as in SUDI for example. However, in the case under discussion the pathologist wrote a vitriolic attack on me saying in essence that it wasn't my province to comment on her postmortem report and what was I doing criticising her findings? So I wrote a note in response going through the evidence in the case in detail and discussing what is known about the adverse effects and overdose toxicology of amisulpride, and pointing out that in my opinion the pathologist should not have placed so much emphasis on the laboratory findings in the first place. This is a double-edged sword of course, as without all the fuss the laboratory error would not have been discovered. This being said, a fatal arrhythmia precipitated by amitriptyline, amisulpride, or by both drugs in combination, could not be ruled out.
As a final straw, Professor Bernard Knight was quoted as saying on the one hand that the investigation of fatal poisoning is a collaborative investigation between the pathologist and toxicologist, and on the other that it is the pathologist rather than the laboratory toxicologist who should provide the final opinion upon the proximate cause of death (her emphasis). Professor Knight 52 actually gives a much more balanced view and emphasises many of the pitfalls that may be encountered in the interpretation of results, but I still beg to disagree with the conclusion that the pathologist has to be pre-eminent because only he sees the whole picture. Many clinical pathologists, and indeed many forensic pathologists, have only limited knowledge of toxicology and therapeutics, especially in specialist fields such as mental health, hence in my opinion to leave the final interpretation of results to them alone is quite simply asking for trouble. The investigation has to be a collaborative effort among the professionals involved under the auspices of the commissioning authority, be it police, coroner, or other body.
Conclusions
I often get the impression that there has been no one in a case who has seen things through from beginning to end and who has the required training and experience to direct the investigation, as I suspect Sir Thomas Stevenson did at the apex of his career. Perhaps it is because I only get to see difficult cases. Or perhaps toxicology has a low priority/is much more complex these days, or is it simply that the process that should be followed is lost among all the other pressures people face (accreditation/ registration/turn-round-time/cost improvement/career development, etc)? But then many cases I see should have been straightforward and only became “difficult” because of a failure somewhere along the line, as evidenced by some of the cases/hypotheses we have discussed today.
Every case of poisoning or possible poisoning is different. Generic toxicology reports with emphasis on “fatal levels” are I think unhelpful, especially in difficult cases. In Stevenson's day a “hack” was a writer who quickly put together low quality articles or books. I fear we are in danger of moving towards “hack” laboratories where cost is everything and value nothing. I see this every day in the National Health Service. A crucial mistake has been to ignore the value of academia in the process. Research informs, and the research process informs the researcher even more in that it teaches how to evaluate other people's work.
Analyses are not the be all and end all of a toxicological investigation. If someone has died, an analysis can prove exposure and perhaps give an indication of the magnitude of exposure, but the results have to be placed in proper context. Even if sampling was antemortem an analysis does not necessarily prove the effects that the drug or poison had on the victim immediately before or at the time of sampling. Tolerance is one big issue, the mechanism of exposure (how the drug got into the body) is another, and of course with postmortem work there are always additional considerations such as site of sample collection and the possibility of postmortem change in analyte concentration. And then there are the questions of quality and whether a particular analysis and the interpretation placed upon the result are fit for purpose.
It needs a lot of training and experience together with appropriate financial support to produce a rounded analytical/forensic toxicologist. In my opinion we as a country have largely thrown away our heritage in this field through the rush to privatise/cut costs at all cost, a field that we were really the first to bring to wide application in the service of the courts. The (to my mind) ridiculous decision to just simply close the Forensic Science Service is but the latest example of this malaise. I shudder to think what Sir Thomas Stevenson would make of it all. With that thought, I'll close and simply say thank you very much for your attention. (Applause.)
Discussion
You may have seen on one of the police programmes on the television recently or read in the papers that illicit alcohol containing methanol is still in circulation and remains quite a big problem in the UK. Glen's vodka is a brand commonly counterfeited apparently (http://www.watfordobserver.co.uk/news/2005006.fake_vodka_could_blind_you/, accessed 20 August 2012).
But yes, ethylene glycol poisoning is not common here. The Scandinavians as you rightly say and also the Russians see a lot of ethylene glycol poisoning because they have a big problem with alcoholism. They also have a big problem with clozapine in Russia, by the way, because clozapine is freely available and it is used by prostitutes to knock out their victims. It is the modern Mickey Finn there. And of course clozapine is very toxic in clozapine-naive subjects, so while some of them wake up minus their wallets, others finish up in the morgue. 5
There has been much made of the use of hair analysis to give a longer window of detection for drugs such as flunitrazepam, but it is very rare for this procedure to yield useful information. There is always the possibility of voluntary exposure - if such exposure was after the alleged incident then there is the possibility of diffusion into hair from sweat. Nothing is straightforward. In one case where it was claimed that a low concentration of sildenafil (Viagra) had been detected in a hair sample from a complainant where DFSA was suspected, the prosecution was potentially compromised because the laboratory published their results 5 without informing the investigating authority before the case came to court, and anyway I had serious doubts as to the relevance of the findings.
If the investigation had proceeded properly in the first place and the laboratory had been told, “Look for poisons” and perhaps given more of the circumstances of the case, then we might have had a totally different outcome. I've seen this time and time again in this country as well because of this lack of continuity.