
Abstract
Kawasaki disease (KD) is characterized by persistent fever, mucous membrane hyperaemia, cervical lymph node enlargement, exanthema and periungual desquamation. It is seen mainly in children, with <60 cases reported in adults. We present the case, the first to the best of our knowledge, of a 17-year-woman who developed KD during the second trimester of pregnancy and died 47 days postpartum from cardiac arrest due to acute myocardial infarction. The case, the medical history, the clinical outcome and the postmortem findings are discussed, and we review the literature on adult KD.
INTRODUCTION
Kawasaki disease (KD), also termed as acute febrile mucocutaneous lymph node syndrome, was first described by Dr Tomisaku Kawasaki in 1967 as a disease characterized by persistent fever, mucous membrane hyperaemia, cervical lymph node enlargement, exanthema and periungual desquamation. 1 Although initially thought to be a benign childhood disease, it is now recognized that it can be fatal and also that it rarely occurs in adults, with less than 60 adult cases reported in the literature.
The aetiology of the disease remains unknown. It is hypothesized that it may represent an autoimmune disease, an infectious disease, or both.
KD is the primary cause of acquired cardiac disease in children and adolescents in the UK, USA and Japan. 2 It affects 1/25,000 children in the UK and is fatal in one in 50 of those. The severity of the disease relates to the cardiovascular complications, the most common being the formation of aneurysms in the aorta. Other cardiac complications include cardiac tamponade, cardiac failure, myocarditis, pericarditis, coronary artery thrombosis and acute myocardial infarction. Current treatment is with aspirin and intravenous immunoglobulin, the early administration of which has been shown by meta-analyses to reduce cardiovascular complications. 3,4
Forty-one years after the disease was first described, many of the girls diagnosed with KD are now women of child-bearing age and, therefore, there are a few reported cases of pregnancy, and one major Japanese nationwide survey of pregnancy and delivery in patients with coronary arterial lesions caused by KD. 5–9 There is also a case report of KD in a postpartum patient. 10
To the best of our knowledge, there has not been a case of acute KD during pregnancy. We report the case of a 17-year-old who developed KD during pregnancy and died from acute myocardial infarction, 43 days postpartum, after delivering a healthy boy.
CASE REPORT
A 17-year-old adolescent/young woman was referred from a peripheral hospital for further evaluation and ongoing investigation of abdominal pain, putatively secondary to bowel ischaemia and possible antiphospholid antibody syndrome (APS). She was 30 days postpartum, having delivered a healthy son at 33+ weeks gestation.
Seven months previously, when eight weeks pregnant, she developed a persistent dry cough, which was treated as asthma with two short courses of prednisolone 30 mg daily, without improvement; she had no chest pain, dyspnoea or haemoptysis. She had polyarthralgia in her ankles, knees, hands and wrists, but no swelling or erythema. She also had bilateral conjunctival injection without response to antibiotic eye drops. No laboratory tests had been performed at that time.
Five months later, when she was 29+ weeks pregnant, she was admitted locally with pyrexia of unknown origin. On the day of admission, she developed left shoulder tip pain.
Investigations at that stage revealed mildly abnormal liver function tests, repeatedly negative bacterial, viral and tuberculosis cultures and negative serology (rubella immune, past infection with Epstein-Barr virus [EBV] and parvovirus B19, negative for cytomegalovirus and toxoplasma). The erythrocyte sedimentation rate (ESR) was 120 mm/hour, and C-reactive protein level varied between 30 and 110 mg/L. The full blood count and the bone marrow aspirate showed reactive changes. Lupus anticoagulant testing was positive by Dilute Russell viper assay. Anticardiolipin antibodies, antineutrophil cytoplasmic antibodies and the autoimmune screen (gastric, nuclear, mitochondrial and smooth muscle cell antibodies) were negative. A chest radiograph showed mediastinal lymphadenopathy and a transthoracic echocardiogram was normal.
When 33+ weeks pregnant, she underwent a semi-urgent lower segment caesarean section for fetal growth restriction (FGR) and fetal distress, and delivered a healthy boy. Postdelivery, she remained unwell, with ongoing fever (>38°C), persistent tachycardia, nausea, vomiting and non-specific abdominal pains. Computerized tomography (CT) of her chest and abdomen showed no lymphadenopathy, but did demonstrate the presence of free abdominal fluid, several splenic infarcts (confirmed on subsequent histological core needle biopsy of the spleen) and possible thickening of the transverse colon.
A repeat of the lupus anticoagulant test was positive again and so, in view of the possible diagnosis of APS, she was started on enoxaparin 40 mg/day at one week postpartum, increasing to full anticoagulation levels nine days later, when her splenic biopsy result became available. During this time, she received empirical antibiotic therapy with ciprofloxacin and amoxycillin and clavulanic acid. Unfortunately, she developed diarrhoea associated with Clostridium difficile toxin (CDT) positivity, for which she received oral vancomycin. During this period, eating exacerbated the abdominal pain, she lost five kilograms of weight and her serum albumin fell to 23 g/L. She was depressed because of a combination of difficult social circumstances and possible postnatal depression and saw a consultant psychiatrist who prescribed antidepressants. Because of the persistent abdominal pain and the possibility of bowel ischaemia secondary to APS, she was transferred 31 days postpartum.
On admission, she had been afebrile for a week, but her nutritional status was poor, she had ongoing abdominal pain (treated with opiates) and was tachycardic and hypotensive, with moderate respiratory alkalosis. Her blood results were: haemoglobin: 8.6 g/dL, mean cell volume: 76.5 fL, white cell count: 15.1 × 109/L (neutrophilia), platelets: 1026 × 109/L, international normalized ratio: 2.96 and she had normal urea and creatinine levels. A repeat CT angiogram of her abdomen performed showed no evidence of stricturing.
On the second day of her admission, she had diarrhoea and melaena, which was found to be CDT positive again and, thus, she was treated with metronidazole. At that time, the ESR was 120 mm/hour and the anticardiolipin antibody was weakly positive. The extractable nuclear antigen and double-stranded DNA were negative.
An echocardiogram was arranged in view of her persistent tachycardia and hypotension, and showed poor left and right ventricular function, with an ejection fraction of 30%, left ventricular apical hypokinesia and a small pericardial effusion. A working differential diagnosis of either puerperal cardiomyopathy or a microvascular cardiomyopathy related to the APS, was made. Ramipril was started and full anticoagulation continued. On the twelfth day of her admission, she had two episodes of coffee ground vomiting and her haemoglobin fell from 9 to 6 g/dL, so an oesophago-gastroduodenoscopy was performed, which was normal. Two days later, a naso-gastric tube was passed to aid nutrition. On the fifteenth day, she developed nausea, chest pain and sinus tachycardia (144 beats/minute). One hour later, she underwent a pulseless electrical activity cardiac arrest. Despite cardiopulmonary resuscitation and thrombolysis, she was declared dead 50 minutes later.
A postmortem was performed at the request of the Coroner, in accordance with the requirements of the national Confidential Enquiry in Maternal and Child Health.
At postmortem, the heart showed evidence of biventricular hypertrophy, but there was also extensive thinning, reddish-brown discolouration and mottling of the anterior, posterior and lateral walls of the left ventricle over an area of approximately 10 cm. Histological examination showed acute infarction affecting both ventricles, but predominantly the left and the interventricular septum, with the infarcts ranging from <24 hours to several weeks in duration. All the coronary arteries showed marked, diffuse, concentric thickening of their walls without dilation of their lumens, producing maximal external diameters of approximately 0.7 cm (Figure 1). The entire left coronary artery network was occluded by a recent thrombus. The cardiac valves were normal.
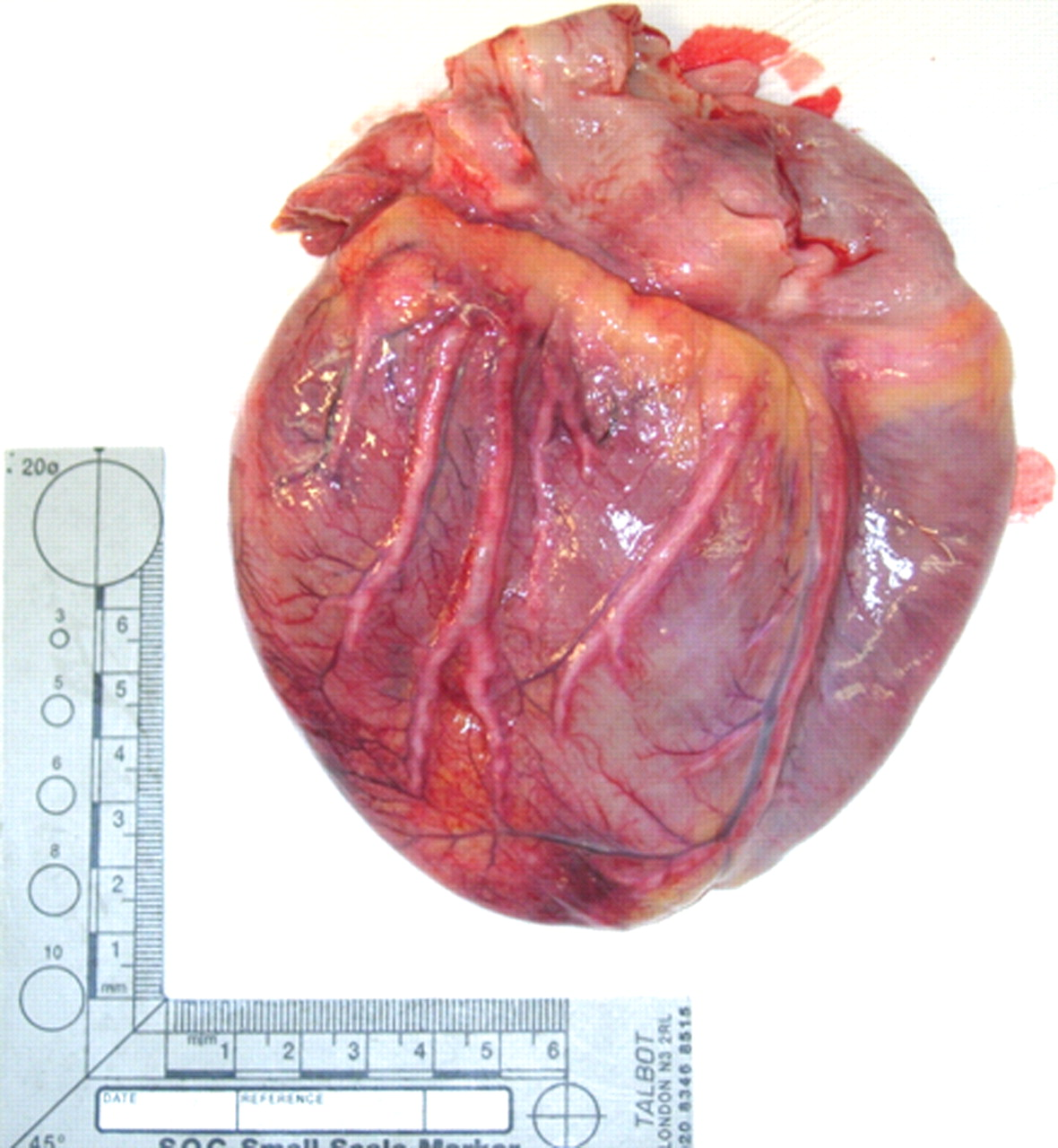
Macroscopic photograph of the external anterior surface of heart at postmortem, showing diffuse thickening of all the coronary arteries
The ascending aorta, distal abdominal aorta, common iliac arteries, renal arteries, carotid bifurcations and the splenic artery showed similar macroscopic thickening. The origins of the major branches from the aorta were not preferentially affected and no aneurysms were present. The pulmonary arteries were normal.
Histological examination revealed an arteritis affecting the large (elastic), medium-sized (muscular) and small arteries throughout the body, sparing the veins and capillaries. The inflammatory cell infiltrate was composed of T-lymphocytes and histiocytes, and was associated with a myofibroblastic proliferation and variable fibrosis (Figure 2). This arteritis was associated with infarcts in the stomach, bowel, spleen, uterine decidua and superficial myometrium, with the splenic and uterine lesions being macroscopically evident. Histology of the liver and kidneys revealed focal hepatic arteritis and renal interlobular and arcuate arteritis, respectively, without associated parenchymal infarcts or inflammation in the samples taken. There was also hepatic congestion and moderate steatosis. Mildly enlarged lymph nodes were identified in the thorax and porta hepatis, which histologically showed areas of infarction.
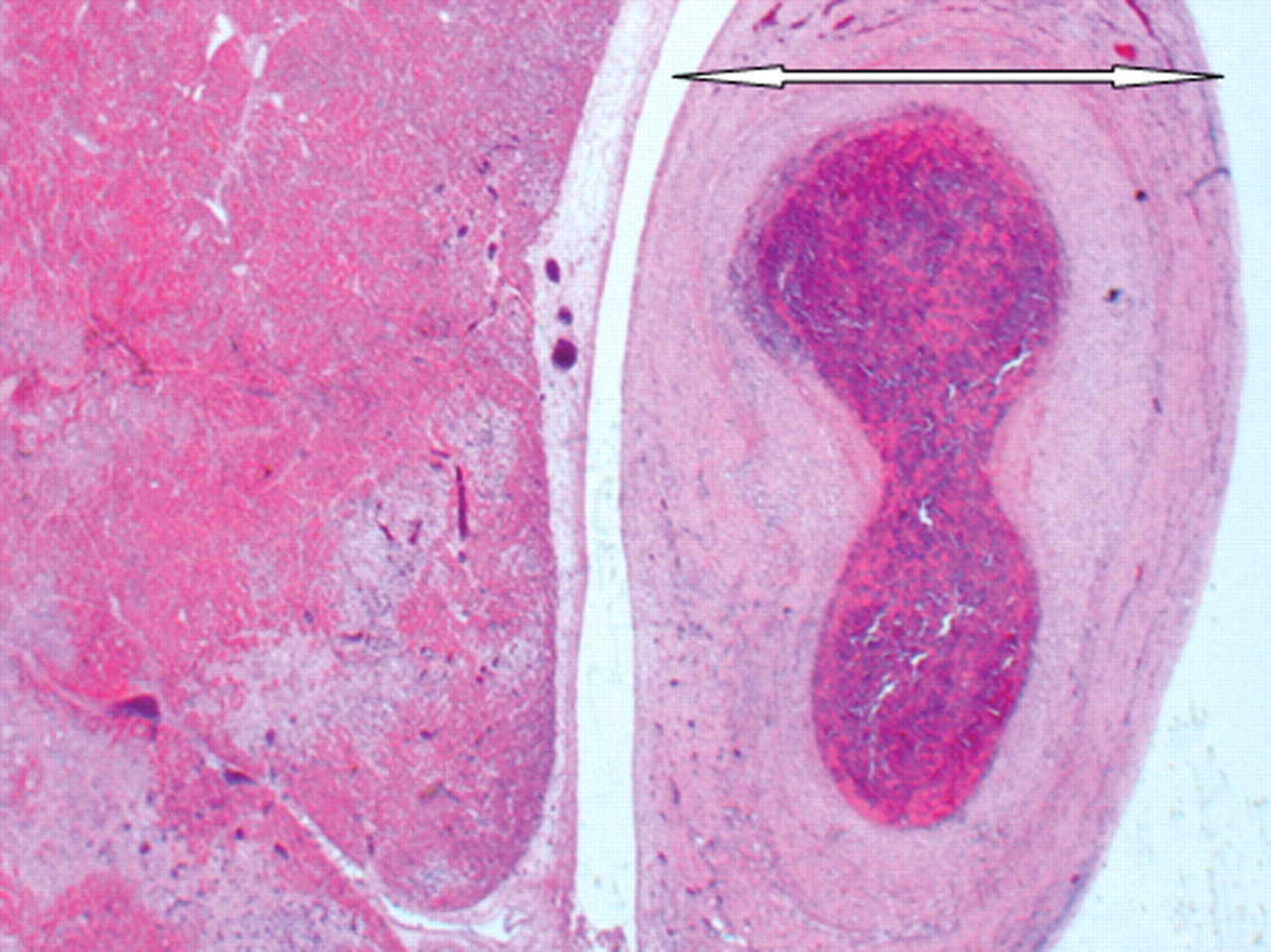
Photomicrograph of the left anterior descending coronary artery (arrowed) showing arteritic thickening and thrombosis, and the adjacent myocardium showing infarction (haematoxylin & eosin ×40)
A diagnosis of the adolescent/adult KD was made on the basis of: firstly, the clinical features at first presentation, namely, pyrexia, arthralgia and conjunctival injection; secondly, the distribution of the arteritis, diffusely involving the arteries of all sizes throughout the body, but with the coronary arteries worst affected, and not the aorta or the origins of its branches; thirdly, the histology of the arteritis, notably lacking necrosis, granulomas or giant cells; fourthly, the involvement of the gastrointestinal tract by the process; and, finally, the lymphadenopathy due to infarction. A correlation of the autopsy findings and clinical findings in this case is given in Table 1.
Postmortem findings and correlation with clinical findings
FGR, fetal growth restriction
DISCUSSION
KD remains the leading cause of acquired heart disease in children and adolescents in the UK, USA and Japan. In many areas it is more common than rheumatic fever and viral myocarditis. 11
KD is nowadays classified as a systemic vasculitis syndrome, affecting predominantly the small- and medium-sized muscular arteries. Although it was initially considered as a benign disease of childhood, it is now known to be the primary cause of acquired heart disease in children and adolescents, which rarely also occurs in adults. Eighty percent of all cases occur in children younger than five years, whereas 71% of adult cases occur between 18 and 30 years. There are no more than 60 adult cases reported in the literature.
The aetiology of the disease remains unknown. Although KD is believed to be caused by an infectious agent in an immunologically susceptible individual, the causative organism remains elusive. 4
KD shows a slight predominance in males, with male/female ratio 1.37–1.5/1 in children, 12 and 1.5/1 in adults. 13 It affects all racial groups, but the Asian ancestry predominates with a 10-fold greater incidence in Japan than in Western countries. In Japan, there are 6000–8000 new diagnoses each year 14 and the mean annual incidence is 90–112/100,000 population, compared with 3.6–3.7/100,000 in Britain and Australia. In the USA, there are 2000 new patients annually, with a mean annual incidence of 6–15/100,000 children <5 years (compared with 50–200/100,000 in children of the same age in Japan), and a peak incidence in children between one and two years. 2,11,15,16
The recurrence rate is very low at 2–3%, 11,15 and the secondary cases are usually in siblings. 16 Familial incidence is reported as 2%. 17
A seasonal distribution of cases has been reported, with preponderance in winter to early spring. There may be epidemic outbreaks, suggesting an infectious aetiology, and major epidemics have occurred in Japan and the USA. Epidemiological studies have identified possible environmental factors, including the use of carpet cleaning, residence near a large body of water, and residence in temperate climates. One study 18 found that the incidence of KD in San Diego correlated inversely with average monthly temperature and positively with average monthly precipitation.
There is some overlap in the clinical features in adults and children, although meningitis and thrombocytosis are more common in children, whereas arthralgia (61.2% in adults versus 24–38% in children), liver function abnormalities (67% versus 10%) and gastrointestinal complications (diarrhoea, abdominal pain, nausea and vomiting) are more often seen in adults. 13 Coronary artery aneurysm occurrence is 20% of childhood cases and the severity of the disease relates to their formation. 13 The overall mortality rate in childhood is <1%, and most deaths occur between 11 and 50 days after the disease onset. 15,19,20 In adults, coronary artery aneurysms occur in 6% and the electrocardiogram changes in 29% of cases. 13 Coronary artery aneurysms are rarely seen in adults with acute KD, in contradistinction to adult survivors of childhood KD. The presence of ANA-positive antibodies is more frequent in adults.
As there is no diagnostic test for KD, diagnosis is based on the clinical criteria and the exclusion of other diseases. Furthermore, because of the limited number of adult cases with KD, current diagnostic criteria have been established on childhood KD and these are shown in Table 2. It should be noted that up to 45% of published cases have an ‘incomplete’ or ‘atypical’ clinical presentation, defined as the presence of <4 major criteria associated with coronary artery involvement. 13 The two most common absent findings are cervical lymphadenopathy and polymorphous erythema. The differential diagnosis includes toxic-shock syndrome (streptococcal and staphylococcal), staphylococcal-scalded skin syndrome, scarlet fever and infection with enterovirus, adenovirus, measles virus, parvovirus, EBV, cytomegalovirus, mycoplasma pneumoniae, rickettsiae and leptospirosis. The differential diagnosis of KD resistant to intravenous immunoglobulin includes polyarteritis nodosa, systemic onset juvenile idiopathic arthritis and malignancy (particularly lymphoma). 4
Diagnostic criteria and other findings in Kawasaki syndrome
CSF, cerebrospinal fluid; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein
KD has two phases: an acute phase, lasting 1–2 weeks and a chronic (convalescent) phase. Usually, the disease is self-limiting and even untreated cases resolve spontaneously after several weeks. 11
Coronary artery lesions usually develop early in the acute phase and only rarely >4 weeks after the disease onset. These early lesions are marked by endothelial cell activation and proliferation, necrosis and adhesion of polymorphonuclear leukocytes to endothelium. These events are accompanied by immune activation, characterized by the increasing levels of circulating cytokines and CD4 and CD8 T-cells, polyclonal hypergammaglobulinaemia, and circulating IgG and IgM immune complexes. 11 Both non-aneurysmal dilatations of the coronary arteries and small aneurysms usually regress after the acute phase, but some moderate-sized aneurysms evolve into stenotic lesions. 21 Severe coronary arterial lesions leading to ischaemic heart disease, including acute myocardial infarction, develop in 2–10%, of whom about one-third are females. 21
In our case, it seems that KD developed during the first trimester of pregnancy with an atypical presentation of fever, cough, bilateral non-purulent conjunctival injection and polyarthralgia, affecting the ankles, knees, hands and wrists, without swelling, erythema or desquamation. Unfortunately, no laboratory data were available from that time.
The pathogenesis of the presence of antiphospholipid antibodies (aPL) in KD, which is described in the literature and was also seen in our patient, is uncertain. aPLs are transiently raised in some infectious diseases and with tissue inflammation and necrosis. It is possible that the exaggerated polyclonal activation of B-lymphocytes that is present in KD, with increases in total serum IgM, IgG and IgA levels, results in the non-specific production of certain autoantibodies, such as aPL. Alternatively, the antigen responsible for mediating KD may cross-react with the membrane phospholipid antigen, resulting in the production of aPL. It could also be possible that the endothelial cell activation seen in KD leads to the exposure of the negatively charged phospholipid surface and thus the formation of aPL. 22
We can find no reference in the literature relating to the impact of pregnancy on the course of KD; we believe this is the first time that the two conditions have been reported to occur together. There is a case report of a patient who developed KD four weeks postpartum, with the formation of coronary artery aneurysms. 10 The authors concluded that pregnancy and the puerperal period did not affect the course of the disease. The patient delivered a healthy boy, albeit at 33+ weeks, with FGR and distress, which may be secondary to uterine infarction due to KD or aPL.
KD is considered to be self-limiting in most cases. Early administration of aspirin and particularly intravenous immunoglobulin has been shown to reduce cardiovascular complications in those with coronary artery involvement. 3,4 Our patient did not receive this, as the diagnosis was not made before death. Although fully anticoagulated, she developed a fatal myocardial infarction, reflecting the inability of anticoagulants to halt thrombosis precipitated by vasculitis. According to a Japanese nationwide survey of pregnancy in patients with residual coronary arterial lesions, secondary to childhood KD, 9 the outcomes of pregnancy and labour in these patients were favourable. The authors concluded that the mode of delivery should primarily be determined by obstetrical considerations, rather than the coronary arterial lesions, and that anticoagulant therapy may not be needed. Our patient delivered by caesarean section without maternal or fetal complication, despite major cardiac pathology and infarction of the uterine decidua and superficial myometrium.
In conclusion, our patient presented with atypical KD in the first trimester. She managed to deliver a healthy boy at 33+ weeks. She received full anticoagulation treatment from the sixteenth day postpartum, but died 43 days after the delivery, from an acute myocardial infarction, secondary to thrombosis on the background of coronary arteritis. Adolescent KD with systemic vasculitis was diagnosed postmortem. Although acute KD is rare in adults and adolescents, its incidence is said to be increasing and, therefore, further studies of its interaction with pregnancy are warranted.