
Abstract
Intrahepatic cholestasis of pregnancy (ICP), also known as obstetric cholestasis, causes maternal pruritus and liver impairment, and may be complicated by spontaneous preterm labour, fetal asphyxial events and intrauterine death. Our understanding of the aetiology of this disease has expanded significantly in the last decade due to a better understanding of the role played by genetic factors. In particular, advances in our knowledge of bile homeostasis has led to the identification of genes that play a considerable role in susceptibility to ICP. In this review we consider these advances and discuss the disease in the context of bile synthesis and metabolism, focusing on the genetic discoveries that have shed light on the molecular aetiology and pathophysiology of the condition.
INTRODUCTION – CLINICAL ASPECTS OF INTRAHEPATIC CHOLESTASIS OF PREGNANCY
Intrahepatic cholestasis of pregnancy (ICP), also known as obstetric cholestasis, presents with maternal pruritus and abnormal liver function in the absence of any other manifestation of acute or chronic liver disease. 1 Clinical and biochemical features of ICP resolve rapidly after delivery. It affects 0.7% of pregnancies in the UK and has a higher prevalence in women of Chilean, Scandinavian and South Asian origin. 2,3
Women with ICP commonly present with pruritus in the third trimester of pregnancy, although severe cases may present in the first trimester. The pruritus is of variable severity. When severe it prevents sleep and may be associated with dermatitis artefacta as a consequence of scratching. Women with ICP rarely have clinical jaundice, although some have dark urine and pale faeces. The biochemical abnormalities associated with the condition include raised serum bile acids and liver transaminases. The extent of the rise in serum bile acids is variable and this is likely to reflect the aetiological heterogeneity of ICP. Serum levels of the liver transaminases, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are also commonly raised in ICP. There is debate about which biochemical test should be used to diagnose the condition. Serum bile acids are more specific to ICP than raised liver transaminases 4 and they are also more valuable for the prediction of pregnancies at risk of fetal complications. 5 The elevation of maternal serum bile acids may occur before clinical symptoms, coincide with them or occur several weeks after the onset of pruritus. Serum bilirubin levels are mildly elevated in a small proportion of cases (<10% of women with ICP in the UK) and serum gamma-glutamyl transpeptidase (GGT) is raised in approximately 30% of cases. 6 Levels of serum alkaline phosphatase help in the diagnosis of ICP as the placental isoenzyme is increased in pregnancy.
Fetal complications that have been reported to occur more commonly in ICP pregnancies include preterm labour, fetal asphyxial events, meconium staining of amniotic fluid and intrauterine death. 5,7,8 Three studies have demonstrated that ICP cases with higher maternal serum bile acid levels (>40 μmol/L in two studies) more commonly have pregnancies complicated by meconium-stained liquor, cardiotocograph abnormalities and fetal asphyxial events, 5,9,10 and the largest study also demonstrated that patients with higher bile acids have higher rates of spontaneous preterm labour. 5
The pathogenesis of the fetal complications of ICP is likely to relate to raised fetal serum bile acids, but the precise mechanisms are not clearly understood. The majority of stillborn infants are of appropriate weight and the evidence to date suggests that the intrauterine death is a sudden event. Several studies have shown an abnormal fetal heart rate 9,11 or arrhythmia 12 in pregnancies complicated by ICP, and in vitro studies of neonatal heart cells indicate that they are susceptible to bile acid-induced arrhythmia. 13 The reason for the increased frequency of preterm labour is not clear, but it has been postulated that it may be a consequence of bile acid-induced release of prostaglandins, which in turn may initiate labour. 14 The increased rates of meconium-stained amniotic fluid 5,8,11,15 may relate to fetal distress secondary to the toxic effects of bile acids or could be a consequence of bile acids stimulating gut motility. 16
Evidence for genetic involvement
ICP has a complex aetiology with a genetic component. A number of lines of evidence support a significant involvement of genetic risk factors. First, the disease shows significant familial clustering. Although large pedigrees are not common, those described show an autosomal dominant sex-limited inheritance pattern. 17,18 Secondly, the incidence of the disease varies between different ethnic groups, suggesting the involvement of population-specific genetic risk factors. For example, the UK prevalence is 0.7%, whereas in Sweden it is 2–3%. 19 Historically, prevalence has been highest in the native Indians of Chile, but recently these figures have fallen. 20 Family studies have provided further evidence of the role of genetic susceptibility in the aetiology of ICP as parous sisters of index cases in the Finnish population have a 12-fold increased risk of the disease. 21 Our own observations support this data in a UK series of 76 caucasian cases in which 14% of parous sisters developed the disease.
The primary phenotype of ICP, i.e. pruritus in association with raised serum bile acids and liver transminases, is indicative of reduced bile flow. Therefore it is necessary to consider the mechanisms and purpose of bile production. Bile consists of three principal components, namely bile salts, phosphatidyl choline and other organic anions. These components are each transported by specific proteins (Figure 1).
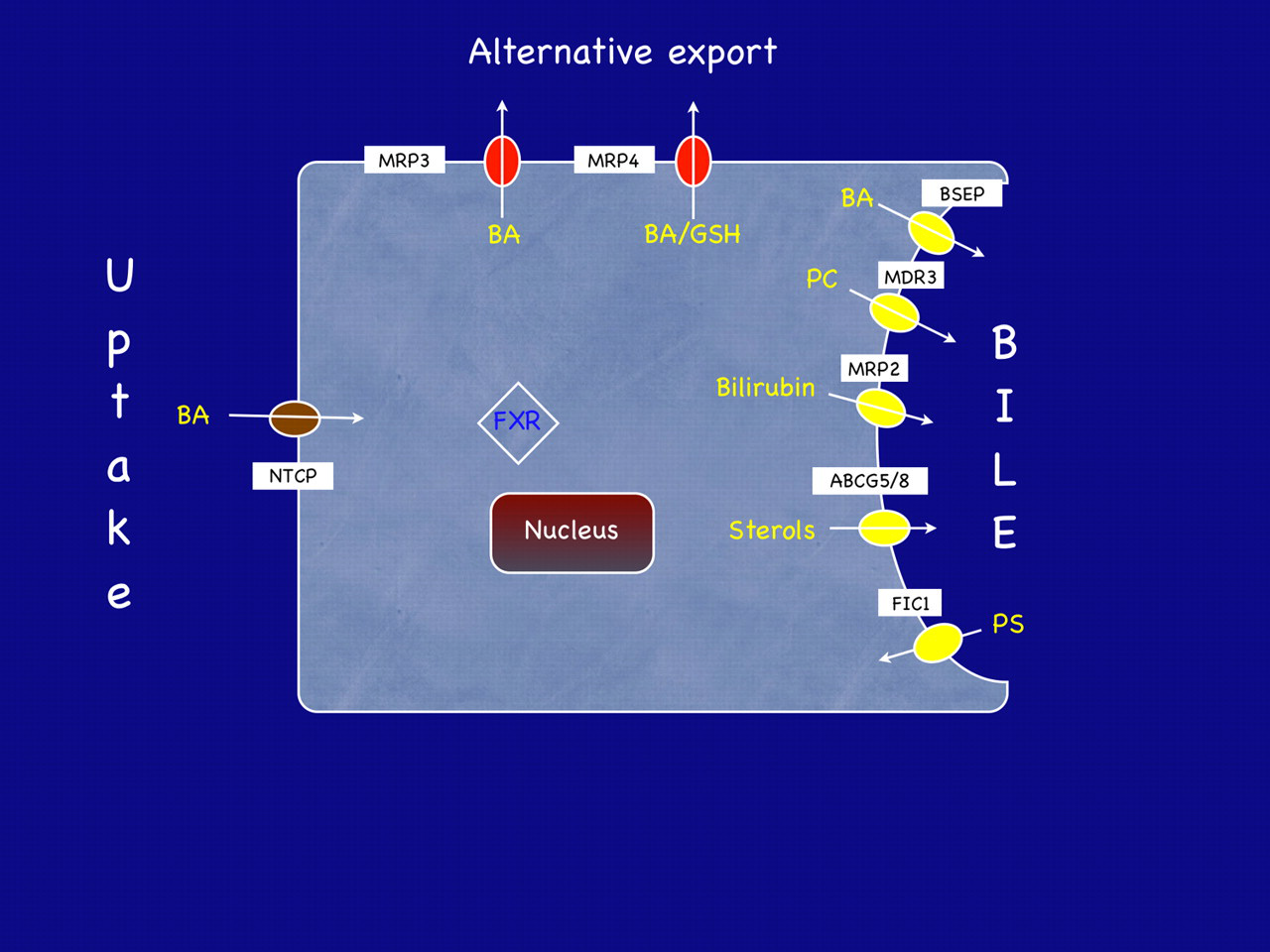
Schematic representation of a hepatocyte showing some of the transporters and receptors discussed in this review. BA, bile acid; NTCP, sodium taurocholate cotransporter polypeptide; MRP, multidrug resistance-associated protein; GSH, reduced glutathione; BSEP, bile salt export pump; MDR3, multidrug resistance protein 3; ABCG5/8, ABC heterodimeric transporter 5/8; FIC1, familial intrahepatic cholestasis 1 protein; FXR, farnesoid-X receptor. MRP3 and 4 are alternative export transporters that can transport bile acids back into the blood. Their role in intrahepatic cholestasis of pregnancy has not yet been examined in detail
Bile acid synthesis and transport
Bile synthesis is a complex process involving the action of at least 17 different enzymes; mostly members of the cytochrome P450 family. In humans the primary bile acids, cholic and chenodeoxycholic acid, are synthesized from cholesterol. Following synthesis they are conjugated to glycine or taurine, thereby forming bile salts to which cell membranes are impervious. The mechanism of intracellular transport of bile salts is not known. Secretion of bile salts following conjugation is an active process against a concentration gradient driven by ATP. A specific transporter, bile salt export pump (BSEP, gene name ABCB11), is responsible for this transport. Other biliary constituents, such as phospholipids, cholesterol and organic ion conjugates, are secreted into the canalicular lumen to form bile. The proteins that contribute to the transport of biliary constituents in the hepatocytes are summarized in Figure 1.
Phosphatidyl choline
Phosphatidyl choline (PC) has a vital protective role in the interluminal space. During bile formation bile salts exported by BSEP form mixed micelles with PC. These complexes serve to protect the lumen epithelium from the toxic and detergent effects of bile salts and, hence, allow their secretion without damage to the surrounding cells. Thus, PC secretion concurrent with bile salts is essential to maintain adequate bile flow. PC is actively secreted into the lumen by the PC floppase MDR3 (multidrug resistance protein 3, gene name ABCB4). In order to maintain normal bile flow, bile salt export by BSEP must be balanced by the presence of PC, flopped into the canaliculus by MDR3.
Other bile components
Bile also serves as an excretory route for bilirubin, drug conjugates and other organic ion conjugates. These organic anions are exported from the hepatocyte by multidrug resistance-associated protein 2 (MRP2, gene name ABCC2). This protein is also present at the canalicular membrane and is responsible for bile salt-independent bile flow.
Following secretion bile is stored in the gall bladder. Upon digestion of a meal, the hormone cholecystokinin acts to contract the gall bladder and release bile into the small intestine. Here bile salts act to emulsify dietary fats, lipids and fat-soluble vitamins. Bacterial modification in the gut results in deconjugation and conversion to secondary bile acids (e.g. deoxycholic acid or lithocholic acid). Bile salts are reabsorbed and are transported back to the liver where the majority are taken up by the sodium-dependent taurocholate cotransporter peptide (NTCP) and the remainder by the organic anion co-transporting polypeptides. In this way bile acids undergo enterohepatic recirculation such that 95% of bile acids are taken up by the liver with the remaining 5% lost in faeces. These are replaced by de novo synthesis as described above.
Other functions of bile acids
As outlined above, bile acids play a key role in facilitating digestion of dietary nutrients. The synthesis of bile acids from cholesterol is the primary route of cholesterol catabolism. In addition, it has recently become clear that bile acids can also act as signalling molecules that influence pathways of cholesterol, lipid and carbohydrate homeostasis, and also influence the immune system. These actions are in part facilitated by the interaction of bile acids with specific nuclear receptors (see below) and also through interaction with the G-protein coupled receptor, TGR5. Thus it is clear that bile acid biology is much more complex than was previously thought.
Homeostatic regulation of bile metabolism
Bile acids are extremely cytotoxic, and, thus, their synthesis and transport is under tight regulation. The nuclear hormone receptor farnesoid-X receptor (FXR) is the principal bile acid receptor in hepatocytes (and in enterocytes) and acts to tightly regulate intracellular bile acid levels within a narrow range. Primary bile acids bind FXR and following heterodimerization with retinoid-X receptor (RXR), the receptor complex translocates to the nucleus and binds to response elements in the promoters of target genes (Box 1). Hence, FXR acts as a master regulator of bile acid levels by sensing intracellular bile acid concentrations and regulating bile acid metabolism and transport. A number of other nuclear receptors and cofactors are involved in bile acid homeostasis, and a comprehensive discussion of the molecular aspects of bile acid metabolism can be found in several recent reviews and the references therein. 22,23
FXR – The principal nuclear receptor in bile homeostasis
When FXR is bound by primary bile acids, it heterodimerizs with its obligate partner, the retinoic acid receptor. This heterodimeric complex translocates to the nucleus and:
Binds response elements (FXREs) in the promoters and induces transcription of genes involved in
Bile formation, e.g. ABCB11, ABCB4, ABCC2
Bile acid conjugation, e.g. bile acid co-A synthetase (BACS)
Bile salt elimination, e.g. sulphotransferase 2A1 (SULT2A1); Binds the FXRE in the promoter of the short heterodimer partner (SHP) gene. This protein product of this gene acts as a promoter-specilic repressor and reduces transcription of genes involved in
Bile acid synthesis, e.g. CYP7A1, CYP8BI, CYP27A1
Bile acid import, e.g. NTCP.
Hence FXR acts through several mechanisms to promote export and to reduce import of bile salts.
ICP genetics
Studies of the molecular genetic aetiology of ICP date back to 1996 when the first DNA analysis of HLA alleles was performed. 24 The subsequent studies of candidate genes/loci for ICP can be broadly divided into those that analysed genes that play a role in biliary transport (summarized in Table 1) and those that do not (summarized in Table 2).
Genetic studies of ICP; genes that influence biliary transport
*Abstract available only in English
Genetic studies of ICP; genes that do not influence biliary transport
*Abstract available only in English
ICP genetics – The PFIC and BRIC loci
Insights into the role of canalicular transporters in ICP first came from the observation that ICP complicated the pregnancies of a subgroup of women whose children had progressive familial intrahepatic cholestasis (PFIC) types 1–3. Affected children in these families have severe childhood hepatic disease and commonly require liver transplants in infancy. The genes mutated in PFIC1, 2 and 3 are ATP8B1 (FIC1), ABCB11 (BSEP) and ABCB4 (MDR3), respectively. It is becoming increasingly apparent that mutations in these genes cause a spectrum of disease ranging from PFIC, to the episodic cholestatic disorder benign recurrent intrahepatic cholestasis (BRIC) and ICP.
ICP-associated mutations in MDR3 were initially identified in a familial case 25 and then a sporadic case. 26 Since these initial findings, a range of other mutations have been described and the role of heterozygous ABCB4 mutations in ICP firmly established, albeit in a limited number of cases (for full details of all mutation reports, see Table 1). MDR3 deficiency syndromes now encompass a range of liver diseases, including cholelithiasis. 27 In addition to the identification of a number of mutations that are inherited in an apparently Mendelian fashion in ICP, a haplotype study in 52 Swedish cases has identified a further role for ABCB4 variation 28 by defining haplotypes across this locus, which are over-represented or under-represented in cases compared with controls. These important initial findings require further investigation in a much larger cohort.
The function of BSEP and the role of ABCB11 mutations in PFIC and BRIC type 2 indicate that variation in this gene could be involved in the aetiology of ICP. This was initially proposed in the Finnish population following analysis of a two SNP haplotype in 57 cases. 29 However, a subsequent study in a larger cohort of 142 ICP cases failed to replicate the initial findings, leaving the role of BSEP open to question. 30
A recent study of the entire coding region of the BSEP gene identified a single potential mutation (N591S) in a cohort of 21 women with ICP. 31 In addition, a polymorphism (c.1331C > T, p.V444A, rs2287622) that may affect hepatic BSEP expression has been suggested to play a role in ICP 31–33 and has recently been reported to be associated with drug-induced cholestasis. 34
Two studies have reported possible mutations in the third PFIC gene, ATP8B1 (FIC1) in UK and Finnish ICP cohorts. 35,36 This canalicular protein is a phosphatidyl serine flippase that is also mutated in BRIC pedigrees that contain relatives with ICP.
ICP genetics – other bile homeostasis loci
Homozygous mutations of ABCC2, the gene encoding multidrug resistance-associated protein 2 (MRP2), cause Dubin Johnson syndrome. Given the location and role of this protein it represents a plausible candidate for ICP. A recent study in the South American population reported an association with a single polymorphism of this gene and ICP. 37 Analysis of several SNPs in caucasians did not show an association. 32 As with the initial haplotype findings for ABCB4, these studies are worthy of follow-up in a much larger cohort.
As described above, the master regulator of bile formation is the farnesoid-X receptor, FXR, which is encoded by the NR1H4 gene. It acts through a number of pathways to regulate bile formation and excretion (Box 1), and two of its well-established target genes are ABCB4 (encoding MDR3) and ABCB11 (encoding BSEP). In a study of UK ICP patients extended to European and mixed-race populations, three genetic variants were identified, which were then demonstrated to have an effect on FXR translation/function. 38
In summary, relatively penetrant mutations in canalicular transporters confer strong susceptibility to ICP while more subtle variation is likely to be responsible for lower levels of risk and to influence susceptibility in greater numbers of individuals. Together with the well-established role for ABCB4/MDR3, there is accumulating evidence for the role of ABCB11/BSEP, NR1H4/FXR and ABCC2/MRP2 in ICP.
A model of the aetiology – role of reproductive hormones
As with other diseases with a complex aetiology, different individuals may have separate variants that will lead to the endophenotype of ICP. It is perhaps worth considering a threshold model whereby individuals have to accumulate a number of ‘insults’ to cross the threshold where cholestasis occurs. The metabolic and hormonal load of pregnancy results in a lowering of this threshold, and genetic variants that reduce transporter activity will further reduce the threshold.
A complete discussion of the role of reproductive hormones in susceptibility to ICP is beyond the scope of this review. However, evidence in accumulating the expression of canalicular transporters can be affected by reproductive hormones and/or their metabolites. For example, in a mouse model 17β-oestradiol administration caused reduced BSEP expression by mRNA analysis. 39 The cholestatic oestrogen metabolite, E217G, has been shown to cause endocytic internalization of BSEP protein, 40 and various oestrogen and progesterone metabolites cause a reduction in BSEP-mediated bile acid transport in isolated Xenopus oocytes. 41 Hence evidence is accumulating that reproductive hormones have a cholestatic effect by acting directly upon BSEP, causing a reduction in available and active protein and hence leading to reduced bile flow.
SUMMARY
Advances in the understanding of bile homeostasis and the genes involved in these processes have shed light upon risk factors for ICP. Future work will focus more upon elucidating the more common population risk factors that taken in isolation make a small contribution, but together account for a much greater proportion of cases than highly penetrant mutations. In addition, examination of the mechanisms of action of reproductive hormone metabolites will allow the molecular aetiology of ICP to be elucidated in much greater detail.
Note
Since the preparation of this manuscript, five further articles have been published which are of relevance to this review:
A large study in a caucasian population (491 cases) has confirmed and expanded the role of ABCB11/BSEP variation in ICP. 64 The authors identified rare occurrences of the common ABCB11 PFIC/BRIC 2 mutations (E297G and D482G) together with the previously identified N591S mutation. In the same study the p.V444A variant allele of ABCB11 was demonstrated to be a significant risk factor for ICP in this cohort. The role of variation in the coding region of the nuclear receptor pregnane-X receptor (PXR, NR1I2) has also recently been studied. In this study of a cohort of 121 caucasian patients, the authors failed to identify a role for coding variation in this gene in ICP susceptibility. 65 Two further studies in Chinese populations have appeared, proposing an association of a CYP1B1 polymorphism with the disease 66,* and the HLA-DR11 allele and the disease. 67 In addition a large family has been described with a spectrum of cholestatic liver disease caused by a homozygous ABCB4 mutation (R788W). 68 In this kindred ICP was present in heterozygous family members during pregnancy.
*Abstract only available in English
Contributorship
The authors jointly prepared the manuscript.
Footnotes
ACKNOWLEDGEMENTS
PD is supported by the Institute of Obstetrics and Gynaecology Trust, The Lauren Page Trust and Wellbeing of Women.
Research in CWs lab is supported by Action Medical Research, The Lauren Page Trust, GlaxoSmithKline, Wellbeing of Women, Women for Women, the BBSRC, the Biomedical Research Centre at Imperial College Healthcare NHS Trust and the Hammersmith Hospitals Trustees.