
Abstract
Congenital adrenal hyperplasia (CAH) is a group of autosomal-recessive disorders caused by a reduced or absent enzymatic activity at one of the stages of adrenal steroid biosynthesis. Prenatal exposure to androgens leads to external genital masculinization of the affected female child. In pregnancy, the provider may be optimizing care for the woman with CAH or targeting treatment to reduce virilization in the affected unborn child. For the affected adult woman the goals of therapy in pregnancy are to prevent adrenal insufficiency, reduce fetal exposure to androgens and glucocorticoids and to avoid damage to reconstructed genitalia. For prenatal therapy for prevention of virilization of possibly affected female children, dexamethasone is used. However, questions remain about the efficacy and safety of exposing 7/8 unaffected children in the first trimester. Prenatal treatment should only be undertaken after careful discussion with the parents of the risks and benefits in an experienced centre or as part of a research protocol.
Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal-recessive disorders caused by a reduced or absent enzymatic activity at one of the stages of adrenal steroid biosynthesis. The result is a reduction in cortisol production, and in 75% of cases, also a reduction in aldosterone production (salt losers). The compensatory increase in secretion of corticotrophin releasing hormone (CRH) and adrenocorticotrophic hormone (ACTH) results in the overproduction of precursors and steroids not metabolized by affected enzyme (see Figure 1). Classification of the different types of CAH is based on the missing enzyme.
Adrenal steroidogenesis pathway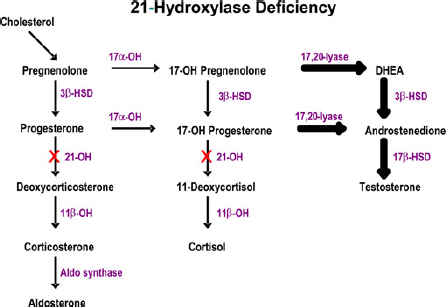
The most common form (>95% of cases), which will be the focus for this review, is due to 21-hydroxylase deficiency resulting from mutations in the CYP21A2 gene. In the classical form excess production of adrenal androgens causes in utero virilization of the external genitalia leading to ambiguous genitalia in affected girls. For boys, or girls not diagnosed at birth, the presenting features include salt-losing crises (failure to thrive, hypovolaemia, shock, neonatal death) or premature adrenarche. The non-classical form is a spectrum of varying degrees of virilization that can be asymptomatic or present as precocious puberty, hirsutism or infertility from childhood up until early adulthood.
Over 120 mutations of the CYP21A2 gene have been identified, accounting for different phenotypic expressions. The disease severity generally correlates with the genetic defect, although there is some non-concordance. The majority of the mutations result in marked loss of function (0-5% of activity) of the enzyme leading to simple virilizing or classical (salt-losing) CAH. 1 The incidence of the severe, classical form is one in 10,000 to one in 20,000 live births, with a carrier rate of approximately one in 60. 2 The less severe form (non-classical) is often the result of a compound heterozygote mutation of two different affected alleles. In one-half to two-thirds of cases, one allele encodes a severe defect and one encodes a mild defect. 1 The phenotype is usually that of the milder mutation.
There are three distinct scenarios in pregnancy:
The woman with classical CAH, who may or may not have a partner who is a carrier or affected;
The woman with non-classical CAH, who may or may not have a partner who is a carrier or affected;
The woman and her partner are heterozygote carriers for CAH which was identified either from having a previously affected child or from genetic screening due to family history.
The Woman with Classical Cah
The woman with classical CAH will usually have been identified as an infant due to ambiguous genitalia. She may have required one or more surgical procedures including cliteroplasty, vaginoplasty and perineal reconstruction and will be on lifelong corticosteroid therapy and a salt-replacing hormone, i.e. fludrohydrocortisone (if a salt loser). Her children will all be carriers of an affected allele; however, none will be affected by CAH unless the father is also affected by CAH or a carrier. If he is a carrier, the children have a one in two chance of being affected.
Glucocorticoid replacement
Glucocorticoid treatment is given to suppress adrenal androgen production and prevent adrenal insufficiency. During childhood, therapy is focused on reducing virilization and optimizing growth. Once final adult height is reached, the goals of therapy are reduction of virilization, ensuring fertility and satisfactory sexual function. 3 Although hydrocortisone is the preferred glucocorticoid for growing children, some clinicians will use prednisone or dexamethasone, especially for adolescent patients as they can be used once or twice daily which may improve compliance. 2 For women likely to conceive, dexamethasone should be avoided unless they have an affected partner (see below). Dexamethasone is not metabolized by placental 11B-hydroxysteroid dehydrogenase and crosses the placenta to the fetus.
Genetic counselling/screening partner
Genetic counselling should be provided to adolescents with CAH to enhance their understanding of the impact of their disorder on their reproductive life. The opportunity to screen their partner (for both male and female patients) to determine if there is a risk of an affected child should be discussed. Genotyping (rather than measurement of 17-OH progesterone) is recommended for determining if the partner is a heterozygote (carrier). 2 Ideally this should be done prior to pregnancy, to ensure time for the couple to understand the implications to their offspring and to explore, understand and agree on their options in pregnancy (see below).
Reproductive function
The fertility rates of women with CAH have been reported to be reduced compared with the general population. The potential mechanisms for impaired reproductive function in women with CAH include overproduction of adrenal androgens and progestins (poorly controlled disease), coexisting ovarian polycystic ovary disease, anatomical outcome of genital surgery and psychosexual development/preferences. 4 The goals of genital reconstruction are to create female appearing genitalia and a functional vagina for menstruation and sexual activity. The timing of the first surgery depends on the severity of the masculinization and parental/provider preference.2,5 Some women will need further surgery in adolescence or a programme of graded vaginal dilation. Confidence and comfort in sexual activity depends somewhat on the cosmetic and functional results of the surgery.
It is important to differentiate fertility from fecundity for women affected by CAH. In one study of 106 women with classical CAH only 24% of the women considered motherhood, of whom 91% achieved a pregnancy, the majority without reproductive assistance. 6 These fertility rates of 0.25 live births per woman are markedly reduced compared with the general population (1.8); however, the pregnancy rates are the same. The lower rates of attempting pregnancy are likely multifactorial and should be explored with all women with CAH. In women with inadequate control of androgen production (manifested by anovulatory menses) or inadequate introitus as the barrier to pregnancy, referral to specialized units is recommended.
Pregnancy optimization and outcomes
In general, the dose of hydrocortisone and Florinef (for salt losers) does not change in pregnancy except at times of stress (i.e. severe hyperemesis, labour and delivery). Although in the non-pregnant state testosterone, androstenedione and 17-OH progesterone levels may be used to monitor therapy, in pregnancy the targets are not clear as 17-OH progesterone, androstenedione and total testosterone levels will increase in pregnancy. Some clinicians use bioavailable testosterone levels, whereas others follow clinically. 7 High levels of progesterone may compete with Florinef for the mineralocorticoid receptor; however, the dose does not need to be altered unless there is clinical evidence of inadequate replacement (based on serum electrolytes, blood pressure, symptoms of deficiency). 2 One concern of poorly controlled disease is that increased maternal androgens may cross the placenta and affect the fetus. Fortunately androgens are metabolized by placental aromatase to estrogen, preventing fetal exposure. 8 Placental aromatase can become saturated in severe maternal non-compliance, causing masculinization in the fetus, but this is very rare. 9
Women with CAH are more likely to need a caesarean section especially in those with more extensive reconstruction surgery.9,10 In one case series, 10 of 13 women with classical CAH had a caesarean section. 9 There are no reports of significant increases in hypertension or diabetes during pregnancy.
The Women with Non-Classical Cah
Women with non-classical CAH may have been identified through investigations for accelerated bone age/premature adrenarche, anovulation/hirsutism or after having a child diagnosed with CAH. This milder form of CAH is associated with 20-50% of residual 21-hydroxylase activity. They do not have significant cortisol deficiency and are not salt losers. The diagnosis is made by measuring 17-hydroxyprogesterone levels after the administration of 250 μg of cosyntropin or through genotyping.
Treatment depends on the clinical presentation and therapy goals. In children with accelerated growth, significant premature pubarche or virilization, glucocorticoid therapy is recommended. 2 In adults with suboptimal fertility glucocorticoids should also be considered.
Genetic counselling is important for women with known non-classical CAH prior to pregnancy. Up to two-thirds of cases are compound heterozygotes with one severe and one mild mutation. 2 Although the mother has mild disease, if her partner is a carrier for classical CAH, the child could have classical CAH. Based on a population carrier rate of one in 60, there is a calculated 1/240 chance of having a child with classical CAH. In one case series, 2/141 mothers with non-classical CAH had a child with severe CAH. In both cases, the mothers’ diagnosis was not made until after the child was born. 11
Known Carrier for Cah: The Pros and Cons of Prenatal Therapy (Table 1)
Preconception genetic counselling
Key discussion points for parents considering prenatal dexamethasone therapy

Role of prenatal dexamethasone
The production of adrenal androgens commences in the developing fetus between weeks 6 and 7 of fetal life.
12
The critical time for sexual differentiation of external genitalia occurs between seven and 12 weeks.
3
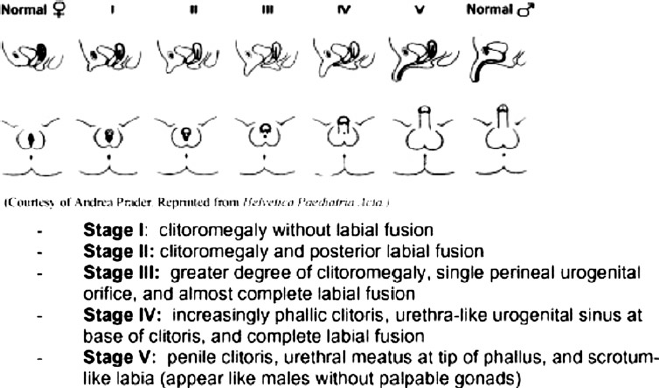
During this time, excess production of fetal androgens can lead to varying degrees of virilization. The severity of virilization may be classified into one of the five Prader stages and can occur in salt-wasting and non-salt-wasting forms of CAH (Figure 2). The fetal hypothalamic- pituitary-adrenal axis is fully functional from six weeks and may be suppressed by exogenous steroid therapy. Dexamethasone readily crosses the placenta and when given to the mother causes suppression of the fetal pituitary-adrenal axis.13,14
Prader score for external genitalia. Used by permission from Nimkarn S, New M
34
Dexamethasone has been studied as a therapy to prevent virilization of the external genetalia of the affected female infant.15-17 However, as genital differentiation occurs early in the first trimester, prior to a time when it is feasible to obtain fetal cells for prenatal diagnosis by amniocentesis or chorionic villus sampling (CVS), treatment may result in the exposure of unaffected fetuses. If the fetus is an affected female, then the dexamethasone is continued until term. In all other situations, the dexamethasone is discontinued (Figure 3).
Diagnostic and treatment algorithm for CAH during pregnancy. Used by permission from New M.
16
Copyright, The Endocrine Society http://jcem.endojournals.org

Use of dexamethasone for prenatal therapy has been widely disputed with opinions based on a small number of case series. A number of factors including efficacy of therapy, maternal risks, fetal risks and long-term outcomes on offspring need to be considered. The ethical dilemma of exposing seven children for the potential benefit of one as well as the risk of invasive procedures such as CVS has led to much debate. Two major publications differ in their recommendations. The Endocrine Society guidelines published in 2010 recommend that prenatal therapy be considered as experimental and pursued only as part of a research protocol approved by institutional research ethics boards. 2 The Lawson Wilkins Pediatric Society and the European Society for Pediatric Endocrinology joint Guidelines also recognize the specialized nature of this therapy and that it is not ‘standard of care’ for a community obstetrician. 18 They recommend this course of therapy be pursued only in an experienced centre with the support of a multidisciplinary team or as a part of a research protocol. Given the varying opinions and ethical factors involved, counselling concerning the potential risks and benefits of therapy prior to pregnancy is crucially important.
Effectiveness of therapy
Data concerning the effectiveness of prenatal treatment of CAH are limited, due in part to the rare nature of the disorder. The largest reported series involved 532 pregnancies assessed for a fetus affected by CAH. 16 In this series, prenatal therapy was initiated in 281 pregnancies with dexamethasone 20 μg/kg/ day in three divided doses started between nine and 11 weeks of pregnancy. Of the 105 affected pregnancies (61 females, and 44 males) dexamethasone was given throughout pregnancy in 49. Of the 25 affected female offspring who received dexamethasone prior to nine weeks, the mean Prader score was 1.0. Normal genitalia was observed in 44% (11/25), 44% (11/25) had minimal virilization with Prader scores of 1 to 2, and 12% (3/25) were virilized with a mean Prader score of 3. In the offspring who received treatment after week 9, the mean Prader score was 3.0. Those who received no therapy were the most virilized with a mean Prader score of 3.75. Lagic et al. 15 reported on the outcomes of prenatal diagnosis and therapy of 44 pregnancies at risk of CAH. Of six affected female offspring treated to term, 50% (3/6) were not virilized, 33% (2/6) had mild virilization with Prader score of 1 to 2, and one child had a Prader score of 2 to 3 requiring surgery in a mother who was poorly compliant with therapy. A recent meta-analysis of 325 pregnancies treated with dexamethasone demonstrated a reduction in virilization as measured by Prader score of affected female fetuses by approximately 2 points on the 5 point Prader scale (weighted mean difference -2.33, 95% CI -3.38, -1.27). 19 The authors concluded that early administration of dexamethasone to female fetuses affected by classical CAH seemed to ameliorate virilization.
Risks of therapy
Maternal risks
Possible maternal side-effects of dexamethasone therapy include excessive weight gain, cushingoid features, hypertension, gestational diabetes, excessive striae, mood lability and suppression of the maternal pituitary adrenal axis necessitating weaning of steroids post delivery. In the largest series of 118 women treated to term with dexamethasone who responded to a self-reported questionnaire, significant differences were found in stria, oedema and mean weight gain (29.7 versus 36.9 lbs), but no significant difference was found for gestational diabetes or hypertension. 16 All mothers who received prenatal dexamethasone treatment stated that they would take dexamethasone again in a future pregnancy. In a smaller series of 44 women receiving dexamethasone therapy to term, no difference in maternal blood pressure or glycosuria were found. 15 A significant difference in weight gain was observed in the first trimester, but this difference disappeared at term. In a self-reported questionnaire, women treated with dexamethasone reported significantly increased appetite, rapid weight gain and oedema than did controls. In this study, however, 68% of women stated that they would decline treatment if offered in a future pregnancy. 15 A meta-analysis has reported an increase in the self-reported frequency of oedema (RR 1.83; 95% CI 1.17, 2.86) and striae (RR 1.62; 95% CI 1.07-2.45). 19 No differences in gestational diabetes were found. There were insufficient data to report on the incidence of hypertension.
Thus, a number of studies have reported maternal side-effects of prenatal dexamethasone therapy, but they appear to be modest. If these complications are severe, consideration to reduce the dose of dexamethasone in the second or third trimester when critical external genitalia formation is complete should be given; however, this may impact the effectiveness in preventing virilization. There are no reported data on risks of maternal osteoporosis and avascular necrosis with prenatal dexamethasone therapy.
Fetal risks
There are significant potential fetal risks that must be discussed with parents.
Short term
The association between high-dose steroid therapy and orofacial clefts in pregnant animals and earlier reports among humans of similar defects has led to the classification of dexamethasone as category C (safety in pregnancy has not been established). 2 A multicentre case control study using data from the National Birth Defects Prevention Study (1143 cases of cleft lip + palate, 628 cleft palate and 4143 controls) examined steroid prenatal exposure used for various indications and orofacial clefts. Thirty-three infants with cleft lip + palate (2.9%), six infants with cleft palate (1.0%) and 72 control subjects (1.7%) reported corticosteroid use from four weeks before through 12 weeks after conception (OR of 1.7 (95% CI 1.1- 2.6)). 20 No reports of cleft lip or palate have been reported as a complication of dexamethasone therapy for prenatal therapy of CAH. In the largest pooled sample to date, a meta-analysis of four studies (325 pregnancies treated with dexamethasone) no increased risk of fetal malformations, stillbirths or spontaneous abortions were found in dexamethasone-exposed pregnancies. 19 In addition, no differences in birth weight and growth parameters have been found between exposed and unexposed offspring.15-17
Another concern is potential adrenal suppression of dexamethasone-exposed infants. Patterns of urinary excretion of steroids have been found to be altered in infants aged 39 days exposed to dexamethasone from weeks 7-9 of gestation. This suggests suppression of fetal adrenal steroids by the dexamethasone even if exposed for only a short time early in gestation. Although this finding has not appeared to cause any clinical problems for newborns, the long-term effects are unknown. 21
Long term
To date, no negative effects of dexamethasone exposure on behaviour or temperament have been reported. 19 In a preliminary study of 26 prenatally dexamethasone-treated children compared with controls, no differences were found in developmental milestones or cognitive development. 16 However, children treated with dexamethasone were found to have greater shyness and inhibition. 22 A recent study by Hirvikoski did not support these results. In their study, 26 dexamethasone-exposed children and 35 matched controls were assessed by parent-completed questionnaires on behaviour and social interactions. 23 No differences were found in parental ratings of behavioural problems or psychopathology. In contrast to the study by Trautman et al., 22 dexamethasone-exposed children were rated by their parents as more sociable than controls. Another study of 174 prenatally treated children and 313 controls involved the completion of four standardized questionnaires by mothers on their offspring. No differences were found in cognitive or motor development. 24 Hirivikoski et al. 25 studied 40 dexamethasone-treated children compared with 35 age- and sex-matched controls using standardized neuropsychological tests. They found dexamethasone-exposed offspring had decreased verbal working memory and rated lower on self-perception of scholastic competence. No difference was found, however, in psychometric intelligence, measures of cerebral lateralization, memory encoding and long-term memory. This group has also recently reported on gender role behaviour of children exposed prenatally to dexamethasone in their cohort of 40 children (mean age 11 years). 26 The dexamethasone-treated CAH unaffected girls did not differ from their controls. The dexamethasone-treated unaffected boys showed a trend towards less masculine behaviours (P = 0.13), but this result was not significant. The authors acknowledged the need for larger studies to further clarify these results.
No long-term effects of dexamethasone-exposed children have been reported, but a clinical trial of these outcomes in addition to neuropsychological outcomes is currently ongoing (Clinical trials Reference number: NCT00617292).
Identification of the affected child
The purposes of diagnosing CAH in utero include gaining information to decide (1) to continue prenatal treatment in affected female fetuses, (2) to discontinue treatment in unaffected female fetuses and all male fetuses and thus reduce potential complications, and (3) to provide ongoing treatment in affected neonates to avoid complications of delayed diagnosis in infancy (e.g. adrenal crisis).
The first successful prenatal diagnosis of CAH was through measurement of 17-hydroxyprogesterone and pregnanetriol levels in amniotic fluid levels obtained through amniocentesis. 27 Hormonal levels are of limited value as virilization of affected females begins much earlier in pregnancy and 17-OH progesterone levels may be falsely low in simple virilizing CAH or in mothers receiving prenatal steroid therapy. 12 Currently, approximately 95-98% of the causative mutation for CAH may be identified by molecular genetic techniques on fetal cells obtained through CVS or amniocentesis. 12 The diagnosis may be missed, however, in a small percentage usually in situations where CAH is caused by undetectable mutations, dropout alleles, or maternal DNA contamination28,29 Although CVS and amniocentesis provide tissue for both definitive diagnosis and sex determination, they are both associated with an increased risk of fetal loss which must be considered in the decision to pursue prenatal therapy.
A promising technique for prenatal diagnosis is the analysis of free fetal DNA from a maternal blood sample. This technique is non-invasive so does not have the risk of miscarriage associated with CVS and amniocentesis, and can be performed as early as seven weeks gestation.30,31 It is only available in a few centres for diagnosis of CAH.32,33 It is used more widely for fetal sex typing and could prevent intervention in women carrying male fetuses, reducing the number of fetuses exposed to unnecessary therapy from 7/8 to 3/8.
Summary
Women with CAH should be counselled in adolescence and again at the transition to adult care about impact of their disease on reproduction. With adequate glucocorticoid replacement, most women who attempt pregnancy will be successful. For women who are not desiring pregnancy, sensitive communication around potentially reversible factors, e.g. comfort with genitalia appearance and adequacy should be undertaken.
For women with an affected partner, prenatal therapy for prevention of virilization of possibly affected females with CAH is promising, especially as methods and accuracy of early prenatal diagnosis increase. However, questions still remain with respect to long-term outcomes of prenatal dexamethasone exposure. A decision to pursue this course of therapy requires careful consideration of the risks and benefits of therapy in an experienced centre or as part of a research protocol.