
Abstract
The aetiology proposed for the development of chronic cerebrospinal venous insufficiency (CCSVI) associated with multiple sclerosis (MS) has been the presence of congenital truncular venous malformations. However, this hypothesis is not consistent with the epidemiology or geographical incidence of MS and is not consistent with many of the ultrasonographic or radiographical findings of the venous disturbances found in MS patients. However, the probability of a venous aetiology of MS remains strong based on evidence accumulated from the time the disorder was first described.
The method used in this review was to search PubMed for all past medical publications related to vascular, venous, haematological, epidemiological, biochemical, and genetic investigations and treatments of MS.
Epidemiological and geographical findings of prevalence of MS indicate the involvement of an infective agent. This review of the venous pathology associated with MS describes a hypothesis that the pathogenesis of the venous disease could be initiated by a respiratory infective agent such as Chlamydophila pneumonia, which causes a specific chronic persistent venulitis affecting the cerebrospinal venous system. Secondary spread of the agent would initially be via the lymphatic system to specifically involve the azygos, internal jugular and vertebral veins. The hypothesis proposes mechanisms by which an infective venous vasculitis could result in the specific neural damage, metabolic, immunological and vascular effects observed in MS. The hypothesis described is consistent with many of the known facts of MS pathogenesis and therefore provides a framework for further research into a venous aetiology for the disease.
If MS does result from a chronic infective venulitis rather than a syndrome involving congenital truncular venous malformations, then additional therapies to the currently used angioplasties will be required to optimize results.
MS as a venous disorder
Paolo Zamboni's and co-workers’ 1–5 recent articles on an association between the disabling neurological disease, multiple sclerosis (MS) and what he has labelled ‘chronic cerebrospinal venous insufficiency’ (CCSVI) have generated unprecedented interest in the new hope of finding an effective treatment for the disease. This hypothesis is so radically different from the aetiological theories proposed in the past that the CCSVI hypothesis has been met with much scepticism by the medical scientists from the more traditional fields such as neurology who have tended to favour an auto-immune pathophysiology as the cause for MS. However, treatments based on these traditional theories, although having some benefit for relapsing-remitting forms of MS, have been notable for their ineffectiveness in preventing the relentless, relapsing and debilitating nature of progressive MS. 6
The aim of this article is to document the known evidence for a venous cause for MS and relate this to published past research into the disease and its treatment, thereby developing a consistent neuro-vascular theory on the pathogenesis of MS including the suspected venous pathology.
Historical aspects of the venous system in multiple sclerosis
In 1863, Rindfleisch noted the consistent location of a blood vessel in the centre of MS plaques. He showed changes in the blood vessels and nerve elements secondary to inflammation and hyperaemia. 7 Charcot 8 soon after established that the lesions in the brain spread from the ventricles outwards. He noted the relationship of the lesions to the cerebral veins but did not make any emphasis on this fact. In 1916 the vein–plaque relationship was clearly described by James Dawson 9 in regard to cerebral lesions. Dawson's description also indicated that the lesions, in the form of cone- or dome-like fingers, start at the lining of the ventricles from where they spread outwards into the cortex. This explanation as to the spread of the plaques became known as ‘Dawson's fingers’. Dawson went on to describe large collecting veins running immediately underneath the ventricular lining (the subependymal veins), which were directly surrounded by zones of gelatinous tissue. The involved vein walls appeared partly homogenized, lacking normal structural architecture and encompassed by distinctly widened perivenous spaces in which residues of minor haemorrhages were found.
In 1931 Gabriel Steiner 10 also illustrated that the lesions in MS spread from the ventricle borders outwards into the brain. He confirmed this finding again in 1962 11 but also noted at that time the formation of splash lesions far away from the ventricles. These splashes now known as ‘Steiner Splashes’ did not form close to the ventricles but did however form close to blood vessels distal to the ventricles.
Regarding spinal cord involvement, it had been observed by Carswell, and shortly after confirmed by Cruveilhier that MS patients had spinal cords with a ‘peculiar diseased state’ where pathological patches spread from the spinal cord's lateral contour to different depths. 12 Cruveilhier's specimens show uneven extension of the patches exclusively from the spinal cord's flanks with the tissue lying closer to the lesion's lateral line of origin undergoing more intense changes. Another of Cruveilier's specimens showed preferential lesion alignment along the spinal cord's lateral and posterior aspects. In contrast, both Carswell and Cruveilier demonstrated that lesions affecting the pons had a different distribution pattern with more compact lesions exhibiting a multicentric pattern of spread. Although having different patterns, both the spinal and pontine lesions suggest focal inflammatory reactions spreading eccentrically as a result of an injurious (possibly infective) agent.
In 1937 Putnam and Adler
13
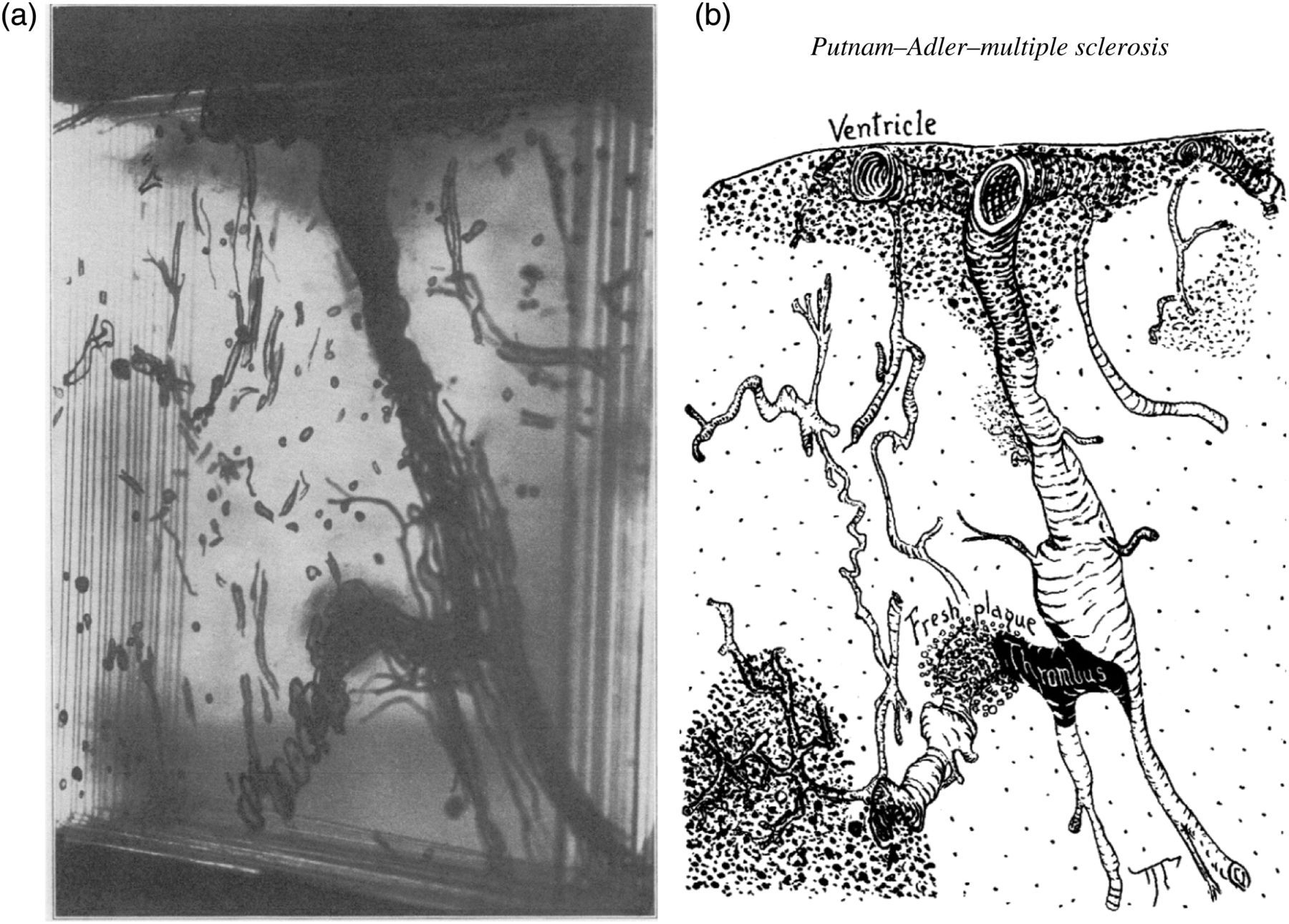
demonstrated that cerebral plaques characteristically spread outwards from the ventricles in a specific relationship to large epiventricular veins, and further to distorted and irregularly distended tributaries of these vessels (Figures 1a and b). They also described several areas of gliosis containing angiomatous plexus of capillaries. This may have indicated either neogenesis or collateralization frequently seen in venous disorders where there is obstruction. With India ink staining of the cerebral vessels they demonstrated how the dilated and tortuous cerebral veins of the MS patient differed from that of the normal patient. The pattern of tissue scarring indicated that the associated veins were considered as pathways for the progressive advance of the disease process into the periphery of the cerebrum.
(a) Glass plate model of multiple sclerosis affected brain stained by Mallory's connective tissue stain to give ready differentiation of myelin, gliosis, vessel walls, red cells, platelets and fibrin. (b) Line drawing interpretation of the glass plate model. Practically the entire wall of the lateral ventricles was lined with gliotic tissue in which lay large veins, many of them surrounded by haematogenous pigment. The sclerotic tissue followed the radial veins for a variable distance, and irregular patches of sclerosis were found at intervals along them. Particularly striking was the irregular, tortuous, congested contour of the main veins (from Putnam and Adler
13
)
In 1965 Fogg 14 also showed that the shape and course of the cerebral periventricular plaques are determined by the course of the great central periventicular venous plexus and their arborizations into the white substance of the brain. Fog concluded that the shape and course of cerebral plaques in MS are determined by the course of the venous system.
An infective agent in MS
The concept of an infectious agent causing MS was first promoted by Pierre Marie. He initially reported a case of MS developing after recovery from typhoid fever. 15 Marie 16 subsequently stated: ‘There is a plainly clear cause of the sclerotic cerebrospinal plaques. This quite effective cause is infection, or a series of infections.’
Much later another French physician/epidemiologist, Paul le Gac, reported his theory on an infectious cause of MS to the Academy of Sciences meeting in Paris 1960. Le Gac 17–20 suggested on epidemiological and serological evidence that many cases of MS were the result of complications from infections with Rickettsioses and neo-Rickettsioses (Chlamydophila) and that MS was the result of the infection affecting the vascular system, together with a disturbance of the nutrition of the nervous tissue affected by the vascular changes; this involved localized anoxia and retardation of the nerve tissue metabolism. He then went on to describe successful treatment of 30 cases of MS with broad-spectrum antibiotics. 20 Most, but not all, of the patients were sero-positive for Rickettsia or para-rickettsioses (Chlamydiacea).
Although Le Gac inferred that many cases of MS were the result of an insidious persistent infection spreading via the vascular system, it was CWM Adams,
21
a neuropathologist, who provided the first indirect evidence of an infective venous vasculitis in MS. By examining 25 autopsy cases of MS he reported ‘for the first time that the cerebral venular wall in MS is the site of lymphocytic infiltration that may at first, particularly in grossly normal white matter, be confined to the vessel wall alone (Figures 2a and b). As the inflammatory process proceeds, the cellular infiltrate appears to spread to the perivascular space and even into plaque tissue. Vein walls then undergo focal intimal hyperplasia, intimal organization and collagenous thickening (Figure 3). Such features suggest a mild expression of subacute or chronic endovenulitis of the cerebral veins. Lymphocytic infiltration of the vein wall alone frequently occurs in the absence of any surrounding perivenular infiltration, and that such intramural inflammatory changes in the vein wall may be located at a distance from plaques in areas of grossly normal white matter or in oedematous (but otherwise normal) white matter. This particular sequence of events strongly suggests that the inflammatory process may start or be first manifested in the vein or venular wall and argues against it merely being a result of brain damage. The usually modest nature of the venulitis counts against a bacterial or viral attack, unless the agent is relatively benign or incomplete in nature.’
21
(a) Lymphocytic infiltration and oedematous onion-skin change in vein wall in normal white matter about 1.5 cm from an active multiple sclerosis plaque, VMT, ×300 (from Adams et al.
21
) (b) Mononuclear and macrophage infiltration in and around a vein wall near an active multiple sclerosis plaque, antimuramidase peroxidase, ×120 (from Adams et al.
21
) Collagenous thickening of a vein wall and organizing endothelial encrustations (arrows) in a chronic periventricular plaque, VMT, ×200 (from Adams et al.
21
)

Zamboni's CCSVI
Zamboni and Galeotti
22
defined the concept of CCSVI as ‘a syndrome characterized by stenosis of the internal jugular and/or azygos veins (IJVs-AZ) with opening of collaterals and insufficient drainage proved by reduced cerebral blood flow and increased mean transit time in cerebral magnetic resonance imaging perfusion study.’ As defined, this condition has been found to be strongly associated with MS. Using duplex ultrasound Zamboni described a protocol whereby the diagnosis of CCSVI could be made. The diagnosis depends on examining the IJVs and vertebral veins (VVs) – the extracranial venous assessment (ECVA) – in the supine and upright positions, and transcranial duplex examination of the intracranial veins. In the supine position in the normal subject, flow through the IJVs is favoured, whereas in the upright position flow through the VVs is increased.
23
Zamboni then proposed five parameters to determine whether CCSVI was likely to be present:
Reflux in the IJVs and/or VVs in upright and/or supine positions; Reflux in the deep cranial veins; High-resolution B-mode evidence of IJV stenoses; Flow not Doppler-detectable in the IJVs and/or VVs; Reverted postural control of the main cerebral venous outflow pathways.
Diagnosis of suspicious extracranial venous outflow required at least two of the five above listed criteria to be fulfilled.
3
This would then be an indication to proceed to selective venography to identify accurately any obstructive stenoses and then proceed to percutaneous transluminal angioplasty.
5
Zamboni has attributed the development of CCSVI to the presence of truncular venous malformations that represent embryologically defective veins where developmental arrest has occurred during the vascular trunk formation period in the ‘later’ stage of the embryonic development. 24 The main dilemma for this model of CCSVI and its claimed association with MS is that it assumes a congenital origin is pivotal to the development of MS which contradicts many of the known facts of MS, in particular those related to epidemiology and geographical distribution. 25 In addition, there is no pathological evidence that these ultrasound and radiologically diagnosed abnormalities are truncular venous malformations.
Epidemiological evidence for an infective cause of MS
The compelling epidemiological evidence for MS as an infection has been extensively reviewed by Kurtzke. 26 Geographically, MS prevalence increases with distance from the equator in both hemispheres. Specifically, prevalence is highest in northern and central Europe (except northern Scandinavia), Italy, southern Australia and northern regions of North America. Middle-risk areas are southern Europe (except Italy), southern USA, northern Australia, northern Scandinavia, the Caucasian sections of South Africa and possibly Central America. Low-risk areas include tropical parts of Africa and Asia, the Caribbean, Mexico and possibly northern South America. 25 Miller et al. 27 found that while prevalence and mortality rates of MS in Australia and New Zealand were strongly correlated with latitude, there was no statistically significant correlation of proportion of Mc/Macs in the phone book (a crude proxy for Scottish ancestry) or frequency of DL2 (an antigen most closely associated with MS) with latitude. They concluded that environmental factors were more likely to explain variations in MS prevalence across Australia.
On the other hand, migration studies have found that groups who migrate from a high prevalence area to one of low prevalence often exhibit higher rates of MS prevalence than the indigenous population. 28 MS among people who migrated as children is usually much closer to that of the native born population, suggesting that environmental factors can moderate the susceptibility to MS. In a study of European immigrants to South Africa, Kurtzke et al. 29 were able to show that natives of high-risk areas are not susceptible to MS acquisition much before the age of 15, and that there is a long incubation period between acquisition and onset of symptoms. The opposite was also found to be true with migration from low-risk areas to high-risk with the low-risk migrants adopting the high-risk incidence of their adopted land. 30
From these epidemiological studies Kurtzke 25,26 concluded that MS was primarily an environmental disease acquired after childhood and that acquisition required prolonged or repeated exposure to the environmental agent followed by a prolonged latent or incubation period between acquisition and symptom onset which he estimated to average 10 years.
If the environmental agent was infective, one would expect some evidence of the occurrence of an epidemic if the virulent infectious agent was introduced to a susceptible population for the first time. Kurtzke and Hyllested 31–33 found evidence of this on the Faroe Islands in the North Atlantic Ocean. There were no cases of MS observed in the Faroes prior to 1943. The Faroes were occupied by 1500 British troops for five years from April 1940. There were then 16 patients with onset of MS between 1943 and 1949. The rate exceeded 10 per 100,000 in 1945. The evidence suggested that MS was acquired only if they were at least 11 years of age at first exposure, and only if the exposure was then for at least two years duration. The location of troop encampments were strongly correlated with the place of residence of all MS patients. Kurtzke concluded that British troops brought MS to the Faroese in the Faroe Islands during 1941–1944. Further Kurtzke claimed that clinical MS was a rare late manifestation of infection with what he called primary MS affection (PMSA) which was a transmissible agent, but the clinical result of PMSA, clinical MS was not transmissible. He hypothesized that PMSA was a widespread (but then unknown) persistent infection of adolescents and young adults which only rarely leads to clinical MS after years of incubation. 25,26,34
Chlamydophila pneumoniae as a cause of cerebrospinal venous vasculitis and MS
There is therefore substantial evidence that MS is primarily a venous disorder that is transmitted by an infectious agent. There have been at least 20 infectious agents associated with MS 35 including human herpes virus 6 and Epstein Barr virus, but the known pathogen that has the greatest likelihood of causing a chronic persistent vasculitis and capable of causing secondary neural injury is Chlamydophila pneumonia. 36 Chlamydial infections are associated with a range of chronic diseases that are characterized by inflammation and scarring and result in significant damage to the host. Chlamydophila trachomatis serovars A to C cause the ocular infection trachoma which results in blindness. Ascending infection by serovars D to K of the female genital tract causes salpingitis which in turn leads to fibrosis, scarring, stenosis and obstruction of the fallopian tubes with the eventual complications of ectopic pregnancy and tubal infertility. 37
The chlamydiae are an evolutionary distinct group of eubacteria sharing an obligate intracellular lifestyle and a unique developmental cycle which begins when infectious, metabolically inert elementary bodies (EBs) attach to and stimulate uptake by the host cell. The internalized EB remains within a host-derived vacuole, termed an inclusion, and differentiates to a larger, metabolically active reticulate body (RB). The RB multiplies by binary fission, and after 8–12 rounds of multiplication, the RB differentiates to EB asynchronously. At 30–84 hours postinfection, depending primarily on the infecting species, EB progeny are released from the host cell to initiate another cycle. 38 It is also now accepted that in addition to this biphasic paradigm of chlamydial development that there is a aberrant persistent phase (intracellular/non-replicating) also known as the cryptic body that represents a critical survival mechanism of chlamydial species and allows the organism to cause a persistent infection with ongoing adverse metabolic effects.
C. pneumonia is a respiratory pathogen that is also endotheliotropic and neurotropic. 39,40 Most people in developed societies are exposed to C. pneumoniae at sometime in their lifetime. Antibody prevalence increases rapidly at ages 5–14, reaches 50% at the age of 20 and continues to increase slowly to 70–80% at ages 60–70. 41
The first study published to find an association between C. pneumoniae and MS was from Vanderbilt University, USA published in 1999. 42 The cerebrospinal fluid (CSF) from 17 patients with relapsing-remitting MS, 20 patients with progressive MS and 27 patients with other neurological disease (OND) were examined for the presence of C. pneumoniae antigen and antibodies to C. pneumoniae. C. pneumoniae was isolated from CSF in 64% of MS patients versus 11% of OND controls. Polymerase chain reaction (PCR) assays demonstrated the presence of C. pneumoniae MOMP gene in the CSF of 97% of MS patients compared with 18% of OND controls. Finally, 86% of MS patients had increased CSF antibodies to C. pneumoniae elementary body antigens as shown by enzyme-linked immunosorbent assay absorbance values that were 3 SD greater than those seen in OND controls. There have since been a number of other studies that have provided evidence of the involvement of C. pneumoniae in MS. 41–53
Chlamydophila pneumonia venulitis pathogenesis – the role of the lymphatic system
The spread of C. Pneumoniae from the lungs to the vasculature has been studied in New Zealand white rabbits by Geiffers et al. 39 C. pneumoniae infection of the lungs results in an interstitial and alveolar pneumonia with bronchiolitis that resolves spontaneously after 2–4 weeks. Histology reveals infiltrates of heterophilic granulocytes and mononuclear cells within the interstitium, alveolar space and bronchiolar lumen. After three days, the granulocytic infiltrates are replaced by mononuclear cells. Sometimes there is a mild vasculitis and perivasculitis within the first three days. Perivascular and peribronchiolar lymphatic hyperplasia is observed from day three until up to eight weeks from initial infection. Geiffers proposed that granulocytes act as a kind of Trojan horse for C. pneumoniae in the early stage of infection, granting access to the alveolar macrophages, which arrive later and can disseminate the pathogen through the lymphatic system.
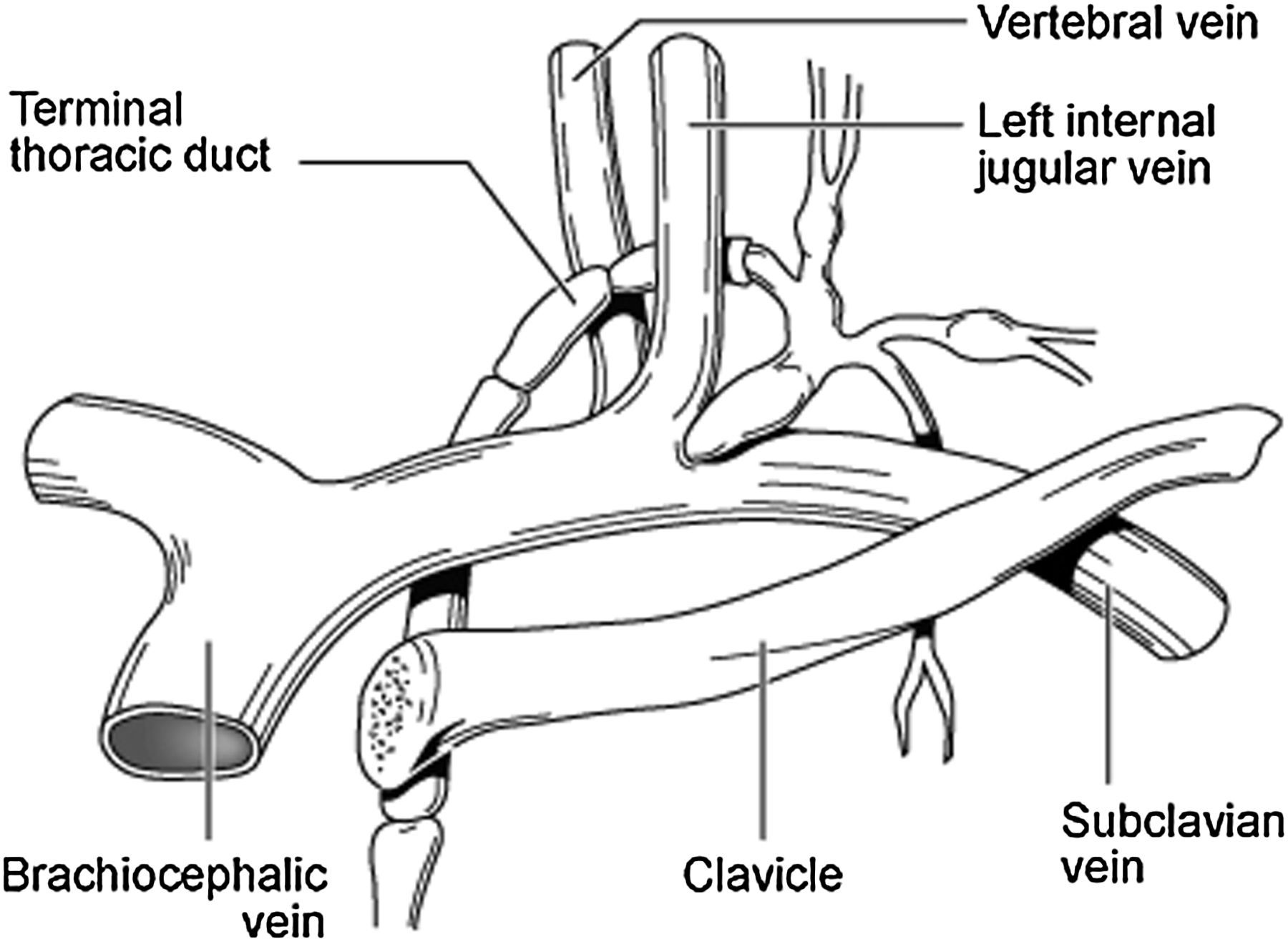
The current author speculates that the organism is then transmitted through peri-hilar lymph nodes within infected macrophages to the thoracic duct and right lymphatic duct. The anatomy of the thoracic duct is depicted in Figure 4. It extends vertically in the chest and curves posteriorly to the left IJV at the C7 vertebral level to empty into the junction of the left subclavian vein, left IJV and left VV (Figures 4 and 5). In contrast, the right lymphatic duct is a short vessel that receives lymph from the right side of the head, neck, and thorax, the right arm, right lung, right side of the heart, and convex surface of the liver and that discharges it into the right subclavian vein at its junction with the right IJV and right VV.
Relative anatomy of the thoracic duct. Note the close association of the thoracic duct to the azygos vein on the thoracic spine. It has been demonstrated in dogs that 22.7% have lymphatico-venous communications between the thoracic duct and the azygos vein
55
The termination of the thoracic duct at the confluence of the subclavian vein, left internal jugular vein and left vertebral vein. Infected macrophages and lymphocytes with C. pneumoniae transmit the infection to the venous endothelium at this site, triggering a creeping venulitis to affect the ophthalmic and cerebral tributaries
Cole et al. 54 have shown that under appropriate conditions pathogenic organisms from infected sites filter through regional nodal barriers of defense and reach the thoracic duct and right lymphatic duct to enter the blood stream and ‘metastasize’ to distant sites. It has also been demonstrated in dogs that 22.7% have lymphatico-venous communications between the thoracic duct and the AZ vein. 55 Infected macrophages therefore have the opportunity to transmit the C. pneumoniae to the venous endothelium through the AZ system in possibly a minority and then at the confluences of the IJV, vertebral and subclavian veins bilaterally. A creeping infective venulitis could then spread slowly and silently distally along the AZ vein in the chest and abdomen, and IJ and VVs in the neck to affect the cerebral, ophthalmic and other intracranial tributaries. Such a mechanism may explain one reported case of recurrent optic neuritis associated with C. pneumoniae infection of the central nervous system in a 12-year-old boy. 56 This patient was finally successfully treated with a combined antibiotic protocol for C. pneumoniae remaining serologically negative and symptom free after six years.
This theory of pathogenesis is also consistent with the findings of Sriram et al. 57 whose work with clinically isolated syndrome suggested that CNS infection with C. pneumoniae occurred early in the course of the disease rather than as an end result of MS. A more recent study of C. pneumoniae immune complexes by Parrat et al. 53 also concluded that infection occurs early in the course of MS disease but that this infection may become chronic.
HSP60, chlamydophila and immune cross-reactivity – the immune link
Heat shock protein 60 (Hsp60) is described in molecular biology as a Group I chaperonin highly conserved during evolution with essential roles in cells and tissues. In eukaryotes, Hsp60 is usually described as a mitochondrial molecule that works with its co-chaperonin, Hsp10, to assist in the correct folding of other mitochondrial proteins. Hsp60 proteins are highly conserved in evolution and, therefore, those of eukaryotes and prokaryotes share numerous identical amino acids. This high similarity in primary structure implies common antigenic sites that elicit and react with cross-reactive antibodies. Exogenous Hsp60 from a microbe can therefore illicit an immune response in humans, a response that although directed primarily against the microbial Hsp60 also reacts with the endogenous Hsp60, providing a link between infection and development of ‘auto-immune’ disease. 58
Hsp60 is primarily a mitochondrial protein but when a cell is stressed it can appear on the cell membranes surface (sfHsp60), where it appears to act as an immune signaling mechanism leading to activation of an antitumour T-cell response. The expression of sfHsp60 positively correlates with the triggering of apoptotic phenonema. 58 Anti-Hsp60 antibodies have been found to occur in a number of systemic auto-immune disease-associated vasculitides, such as Takayasu arteritis, polyarteritis nodosa, Wegener granulomatosis and systemic lupus erythematosus. 59 Humoral immune reactions to bacterial Hsp60 from C. pneumonia and Eschereschia coli have been suggested to be involved in the process of vascular endothelial injury during atherosclerosis pathogenesis. 58
During persistent infections Chlamydia species produce large quantities of Hsp60, which have nearly identical amino acid sequences to human Hsp60. Chlamydial Hsp60 is released from cells infected with the organism and anti-chlamydial Hsp60 antibodies are produced by the host's immune system. In turn, these antibodies recognize surface Hsp on the infected stressed cells and consequently cross-reactivity causes cell lysis and organ damage. 58 This therefore is one explanation for the immune response which occurs in MS, accounting for inflammatory changes observed both in the venous wall and in the surrounding neural tissue.
Chlamydophila infections and host cell energy depletion – the metabolic consequences
Intracellular Chlamydial survival is dependent on active mitochondrial and nuclear function of the host cell. Replicating Chlamydia are always found in close proximity to mitochondria and possess an ATP transport system that imports ATP from the mitochondria and exports ADP. This in effect causes host cell energy depletion and significant metabolic consequences. 60 The biosynthesis of haem occurs in all human cells and is an energy-dependent process which is adversely affected by depletion of host cell energy. Haem synthesis is a series of irreversible biochemical reactions, some of which occur in the cell mitochondria and some in the cytoplasm. The intramitochondrial reactions are mainly oxidation–reduction while those in the cytoplasm are condensation and decarboxylation. Porphyrins and porphyrinogens are formed in the process to produce the end product haem. The porphyrias are consequences of any impairment of the formation of porphyrinogens or in their transformation to haem. Each of the genetic porphyrias is linked to a deficiency in the haem biosynthesis pathway.
Gajdos 61 demonstrated that inhibiting ATP synthesis resulted in excess porphyrin formation or secondary tissue porphyria. ATP depletion impairs the return of coproporphyrinogen into the mitochondria for the final steps in haem biosynthesis leading to accumulation of porphyrins within the cell. The clinical result of this accumulation is a tissue-specific secondary porphyria. Secondary effects of the tissue-specific porphyria include oxidative damage and the accumulation of Fe within the cell and in the extracellular milieu. This may therefore be an additional mechanism to explain the perivenous haemosiderin deposition in MS patients in addition to the vein wall damage described by Adams 62 and the venous reflux proposed by Zamboni. 1
Dietary management of MS has been described by Swank and Dugan. 63 It is of interest that the major aspects of his recommended diet including low saturated fat, avoidance of red meat and high proportion of complex carbohydrates also form the basis of the dietary management of porphyria.
Respiratory infections and vitamin D deficiency – the geographic link
It is now well accepted that the geographic prevalence of MS increases with increasing latitude from the equator. 26,64 An inverse relationship between solar radiation and prevalence of MS was first described in 1960 65 and confirmed by an Australian case-control study in 2003. 66 A recent comprehensive review of MS prevalence confirmed a statistically significant positive association between MS prevalence and latitude globally. 67 The presence of a positive gradient in Europe and Australia strongly supports a role for environmental factors which vary with latitude, the most prominent candidates being ultraviolet radiation and vitamin D.
Vitamin D apart from its classical role in the calcium homeostasis pathway is believed to have an important role in the immune system and endothelial barrier function. 68,69 Vitamin D plays a role in the defence against infective diseases through several mechanisms. 70
The first level of defence is the physical barrier of epithelial cells in the skin, gut, respiratory and urinary tract, which protect from invasion by injury or infection. The active hormone 1,25(OH)2D is important in upregulating genes via the 1α-hydroxylase enzyme which then encode proteins for tight, gap and adherens junctions. Second, vitamin D is a potent stimulator of antimicrobial peptides. The production of cathelicidin and some defensins is dependent on sufficient circulating 25(OH)D. The local production of 1,25(OH)D, the active hormone in various tissues has the ability to induce expression of cathelicidin in bronchial tissues. Local injury or infection in most epithelial sites results in the expression of cathelicidin. 70
The generally accepted immunological mechanism to explain the inverse relationship between vitamin D and the prevalence of MS is that ultraviolet radiation can attenuate T helper cell type 1 mediated immune responses. 71 However, this is a limited view based on the assumption that MS is primarily an immunological disease. An alternative hypothesis is that vitamin D deficiency is associated with MS indirectly through the effect of vitamin D deficiency on respiratory tract infections and subsequent dissemination of the infection to other systems.
Both respiratory tract infections and the occurrence of asthma have been associated with vitamin D deficiency. 72–74 In a population-based study, people with MS were more likely to have asthma prior to MS onset. 75 The same researchers, in another population based study, reported that higher sun exposure during childhood and early adolescence was associated with a decreased risk of MS. There was however no association between sun exposure in the decade before onset of MS. 66 This finding is inconsistent with the hypothesis that vitamin D deficiency is implicated through a detrimental effect on the immune system that is thought to be the origin of the pathology in MS. Instead, the findings are totally consistent with the hypothesis that clinical MS is the uncommon end result of a respiratory infection contracted in early childhood or adolescence that remains clinically dormant during a silent prodromal phase. This latent phase occurs within the cerebrospinal venous vasculature following dissemination from the closely associated lymphatic system as explained previously. A multitude of triggers can result in the initial clinical symptoms which then herald the onset of this progressively debilitating neurological disease.
The implications of chronic infective venous vasculitis of the cerebrospinal circulation
The theory described above has significantly different features to that proposed by Zamboni and termed CCSVI. First, the venous disease will be progressive and parallel the severity of the neurological symptoms, whereas with Zamboni's CCSVI the congenital malformations would be fixed and constant from the time of initial diagnosis of MS. Independent recent ultrasound studies using Zamboni's ultrasound protocol for the examination of the extra and intracranial venous circulation have failed to provide evidence of the presence of the truncular venous malformations causing cerebrospinal venous insufficiency. 76,77 These studies did confirm abnormalities in the cerebrospinal venous circulation of MS patients, but the abnormalities were not in accordance with the criteria determined by Zamboni for the diagnosis of CCSVI. Both studies inferred that blood volume flow measurements may give more valuable information in assessing the state of the cerebro-cervical venous system than Zamboni's criteria.
Second, multiple abnormalities are likely to be found in both proximal and distal segments of affected veins in the venulitis pathogenesis contrasting with the focus on the proximal valves in the CCSVI pathogenesis. The chronic infective venulitis can also result in complete occlusion or long annular stenoses of the affected veins (Figure 6). Due to the importance of the thoracic duct in the distribution of the infection to the venous system, the left IJV will be more frequently affected than the right and will tend to be more severely affected. This trend has been reported by several CCSVI researchers.
22,78
The vascular pathophysiology of the venulitis theory is then based on multiple obstructions causing impaired flow and resultant collateralisation rather than the presence of reflux causing venous hypertension. Further, the venulitis hypothesis has solid histological evidence as observed by Adams.
21
The observed histological changes are unlikely to be the result of venous reflux, but more likely to be due to an inflammatory process.
Selective venography of the extracranial neck veins showing a long annular stenosis of the distal (cranial) segment of the left internal jugular vein with collateralization through the intracranial sinuses and vertebral system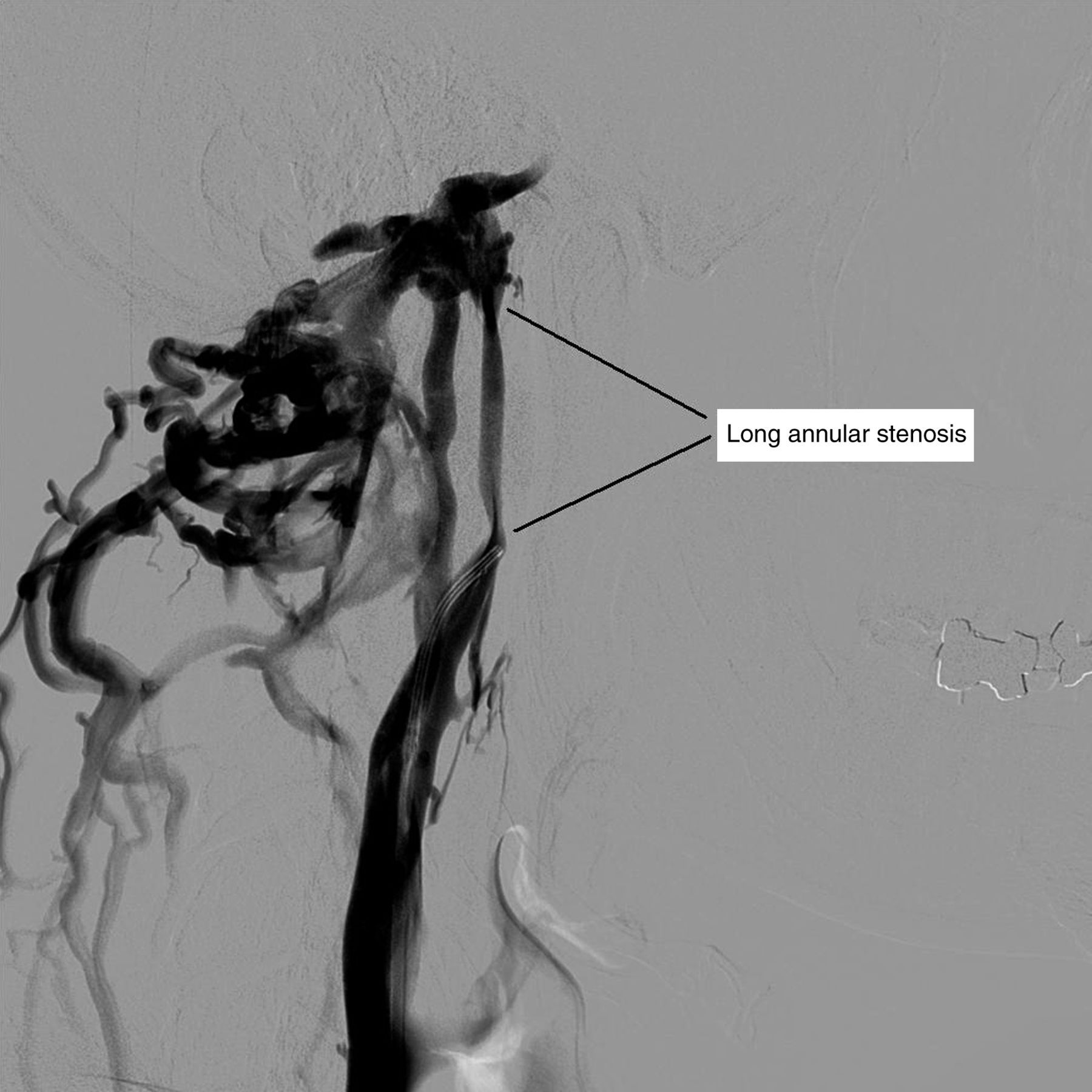
Third, according to the venulitis theory, the stenoses are an end result of the progressive persistent venulitis caused by the chronic infection rather than the primary cause of the neurological damage. The neurological damage is the result of the metabolic and immune processes triggered by the infection. The stenoses may very well have an adverse effect on cerebral blood flow due to venous obstruction, possibly contributing indirectly to decreased cerebral perfusion as described by Zamboni et al. 79 This creates the vascular symptoms of MS such as fatigue. 80 Reversal of the decreased cerebral perfusion may be the mechanism for virtually immediate improvement in symptoms such as fatigue, brain fog and cognition reported by patients following successful angioplasty of stenoses. 80 However, the stenoses and occlusions are not the cause of the MS, they are, along with myelin damage, a result of the infective venulitis.
Finally, the iron deposits found in MS lesions, according to the infective venulitis theory, are the result of metabolic effects from secondary focal porphyria rather than venous reflux proposed by the CCSVI theory. This explains the relatively mild nature of these deposits compared with that found in lower limb chronic venous hypertension and is more consistent with Adam's finding that the haemosiderin deposits occurred in only 30% of MS brains that were examined. 62
Hence, the management of the venous disease associated with MS will be optimized by a multifaceted approach directed at both correction of significant stenoses of the extra-cranial venous outflow and amelioration of the venulitis, which on current evidence is most likely caused by a chronic persistent infection with an organism such as C. pneumonia. Optimum management of MS will then involve a complex holistic approach including optimal antibiotic therapy possibly over a prolonged period, minimally invasive angioplasty of significant stenoses, dietary and nutritional management of metabolic effects including but not limited to vitamin D deficiency and secondary focal tissue porphyria, and finally limited use of immunomodulating drugs at appropriate stages of the disease.