
Abstract
Systemic mastocytosis (SM) is a rare, clonal disorder of the mast cell (MC) and its precursor cells. It is characterized by proliferation and accumulation of MCs within various organs, most commonly the skin. The clinical course is variable with indolent or smouldering and aggressive forms being described. We report the case of a middle-aged male patient with smouldering SM presenting with atypical recurrent life-threatening crises. The patient reported a 19-year history of chronic symptoms. The patient had four inpatient stays due to atypical life-threatening crises, during which he has shown end organ damage (cardiac and renal). With each crisis the patient reported acute symptoms. The management of each of these episodes was complex and made more challenging by the patient's longstanding history of hypertension and ischaemic heart disease. In short, SM can present with unexplained life-threatening crises which can be confused for an infectious disease being acute in nature.
Introduction
Systemic mastocytosis (SM) is characterized by multifocal infiltrates of mast cells (MCs) in visceral organs with or without skin involvement.1–5 A particular category named ‘smouldering mastocytosis’ was introduced into the mastocytosis consensus classification in 2001. 6
Case details
Our patient is a 56-year-old man with smouldering SM. His disease was diagnosed in 2008; however, on careful review of his history, it is likely that he has had this disorder for many years. The patient reported a 19-year history of chronic symptoms, such as exhaustion, bone pain, headache, anxiety, skin rash and itch, flushing, chest pain and nasal stuffiness. In addition, liver function tests had been deranged on a number of occasions. The patient had four prolonged inpatient stays (up to three weeks) prior to diagnosis (January 2007, September 4 2006, February 1998 and September 1994) with life-threatening crises. In retrospect these were likely related to mediator release during which he experienced organ damage in heart and kidneys, resulting in electrocardiogram changes and renal failure. With each new crisis, the patient presented with acute symptoms including fever, severe headache, intractable vomiting leading to dehydration and hypotension, acute kidney impairment/failure, cardiac ischaemia, throat pain and difficulty in swallowing, worsening bone and joint pain, a marked increase in liver function tests and abdominal pain. The management of these episodes was complicated by the patient's longstanding history of hypertension and ischaemic heart disease. These episodes were initially managed in an Infectious Disease Unit as it was assumed that they represented occult infection. No organisms were isolated. Since diagnosis the patient has had a further four acute Haematology Unit admissions. These were managed aggressively with intravenous fluids, intravenous corticosteroids, antihistamines and antiemetics. This strategy has resulted in shorter (4–7 days), less severe episodes and reduced end organ compromise (for example renal function was maintained with the above strategy and did not deteriorate as it had during previous acute admissions).
Between crises, laboratory investigations are unremarkable with normal full blood count, white cell differential and blood film, calcium and phosphate levels, urea and electrolytes and liver function tests. At the time of diagnosis, spinal radiographs showed diffuse sclerotic change. Superimposed on this, there were several ill-defined lucencies, with a peculiar low attenuation lesion within the L2 vertebra body, the latter with a densely sclerotic rim. The skeletal findings were thought to reflect diffuse and focal manifestations of mastocytosis. Bone density scanning revealed no evidence of osteoporosis.
Bone mrow biopsy confirmed SM (Figure 1). Sections stained for tryptase and CD117 showed a marked increase in MCs with abnormal clustering, consistent with the diagnosis (Figure 2). Serum tryptase (a marker for MC activation) was elevated at 100 μg/L (normal 2–14) and has ranged between 100–164 μg/L over 20 months review. The peripheral blood and bone marrow were positive for the activating KIT D816V mutation, using both allele-specific competitive polymerase chain reaction (PCR) (sensitivity 0.1–1%) and pyrosequencing (sensitivity 10%). Fusion gene comprising of Flip1-PDGFRA was not detected by nested reverse transcription-PCR. The patient was commenced on nilotinib 400 mg daily as cytoreductive therapy, but by six months had shown no response by serial tryptase assays.
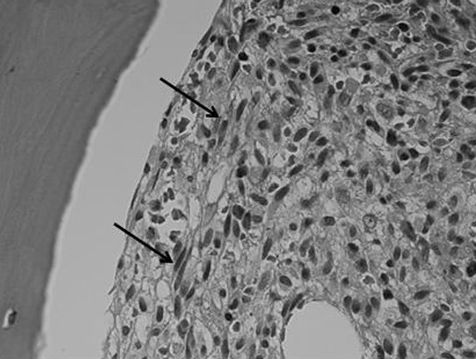
Haematoxylin & eosin bone marrow trephine, ×100 oil immersion. This image demonstrates a focal infiltrate of spindle-shaped abnormal MCs (arrows). Spindling is particularly obvious in the immediate peritrabecular area
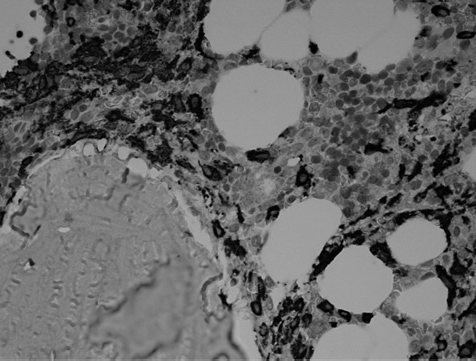
Stained for tryptase by immunocytochemistry, bone marrow trephine, ×100 oil immersion. This image highlights the aberrant MC population, which stains strongly for tryptase (dark stained cells in image). Note abnormal clustering of MCs and the peritrabecular distribution
Discussion
We describe a case of indolent SM with a high MC burden, as evidenced by bone marrow biopsy as well as high levels of surrogate disease markers, including tryptase. The trigger(s) for the life-threatening crises experienced by this patient are still unknown.
Indolent SM is a rare disease that may go unnoticed or may be misdiagnosed, as was the case in this report. For many years, the patient travelled widely with his occupation. On return from abroad in 1994, the patient was admitted to the Infectious Diseases Unit with a proven diagnosis of cholera. On this occasion he had given a nine-day history of loose stool without any associated systemic upset and not associated with blood or mucus per rectum. However, in the 48 hours before admission he had been feeling generally unwell with lethargy, calf muscle pain, headache, burning eyes and nausea, and vomiting. Subsequent episodes of acute illness were then thought to be infectious in nature, though no pathogen was ever isolated. In retrospect these episodes are now considered to have been crises of SM. Failure to make a prompt diagnosis of SM is due to the non-specific nature of many of the symptoms.
The disease is a clonal disorder of the MC characterized by the proliferation and accumulation of MCs within various organs of the body, most frequently the skin. Interaction between the cytokine stem cell factor and its cognate receptor, c-kit (KIT), plays a vital role in regulating MC growth and differentiation. 7 Activating mutations of the c-kit proto-oncogene play a causal role in the pathogenesis. The clinical symptoms and signs are owing to the buildup of these clonally derived MCs in diverse tissues, as well as the discharge of MC mediators, including histamine, heparin, prostaglandins, leucotrienes and cytokines. Manifestations range from exclusively cutaneous or indolent systemic disease to systemic disease with an associated clonal haematological non-MC lineage disorder, to aggressive SM, to MC leukaemia or sarcoma at the other end of the spectrum (World Health Organisation classification). The typical skin lesions are urticaria pigmentosa, which are monomorphic, pigmented, maculopapular or nodular lesions in a prevalent symmetrical distribution, frequently indicating Darier's sign (wheal and adjacent erythema developing in a lesion as a result of rubbing).
There is currently no effective curative therapy for SM. In general, for cutaneous and indolent SM, treatment is aimed at symptomatic relief; this is possible with the aid of agents like antihistamines and MC stabilizers such as ketotifen and sodium cromoglycate. 7 The novel agents’ dasatinib, nilotinib and imatinib (tyrosine kinase inhibitors) may be effective for patients with severe symptoms or with aggressive mastocytosis, 8 although these are not of proven benefit. Imatinib, in particular, is ineffective in the presence of a D816V mutation. Nilotinib, however, is a next generation targeted therapy for treatment of patients with PH+chronic myeloid leukaemia in chronic phase or accelerated phase that have previously shown resistance to or intolerance to at least one therapy including imatinib. 9 In a small group of patients, cladribine (2CdA) and interferon alpha, at times combined with low-dose corticosteroids, have been shown to inhibit disease in 20–50% of patients. 10 Trials testing novel agents such as PKC-412, which inhibits broad spectrum protein kinases, and stem cell transplant as a form of treatment are also underway for patients with the aggressive form of the disease.
Conclusion
In conclusion, because of the non-specificity of some of the symptoms, SM can be difficult to diagnose, as was the case in our report. It is important for pathologists and clinicians to be aware that SM can present with unexplained life-threatening crises, which can be mistaken for an acute illness such as one of infectious nature.