
Abstract
β1–6 branching of N-linked oligosaccharides has been correlated with the progression of different cancers. The leukoagglutinins of Phaseolus vulgaris (L-PHA) have been used to study this pattern of glycosylation whose biological significance is incompletely understood. The animal lectin, galectin-3, also binds to structures recognized by L-PHA. To develop a functional tool for the in situ identification of this pattern of glycosylation, human galectin-3 was fused to bacterial alkaline phosphatase (gal3/AP). Gal3/AP recognized both A and B blood group saccharides (B>A) and lactosamine derivatives. Gal3/AP recognition depended at least in part on the N-linked oligosaccharides of different glycoproteins. The presence and distribution of galectin-3 ligands were analyzed in both murine and human normal and tumor samples. Loss of apical expression of galectin-3 ligands was commonly found in carcinomas. Endothelial and inflammatory cells were enriched in galectin-3 ligands as compared with tumor cells; thus, gal3/AP is a suitable tool for studying tumor micro-environments. Comparative analysis of both gal3/AP and L-PHA binding patterns indicated that although similar, these patterns are not identical. The probe developed was useful for several immunoenzymatic assays and will allow the physiological and clinical significance of the expression pattern of galectin-3 ligands to be established. This manuscript contains online supplemental material at http:/www.jhc.org. Please visit this article online to view these materials.
G
Galectin-3 is expressed in a variety of cells including epithelial and endothelial cells (Glinsky et al. 2001; Lin et al. 2002; Mengwasser et al. 2002; Khaldoyanidi et al. 2003), activated macrophages (Dong and Hughes 1997; Kim et al. 2003), and dendritic cells (Swarte et al. 1998; Vray et al. 2004). A potential role for galectin-3 in inflammation has been suggested by experiments using galectin-3 null mice (Colnot et al. 1998; Hsu et al. 2000; Bernardes et al. 2006). In fact, recent evidence suggests that galectin-3 is a pro-inflammatory protein (Colnot et al. 1998; Hsu et al. 2000). It downregulates interleukin (IL)-5 gene expression in human eosinophils (Cortegano et al. 1998), activates NADPH oxidase (Almkvist et al. 2004), stimulates superoxide production from neutrophils (Yamaoka et al. 1995), and promotes monocyte chemotaxis (Sano et al. 2000). In the extracellular space, galectin-3 may act as a de-adhesion molecule, as shown in the interaction of thymocytes and thymic microenvironmental cells (Villa-Verde et al. 2002) and stimulate cell migration (Silva-Monteiro et al. 2007) and cell death (Stillman et al. 2006; Silva-Monteiro et al. 2007). These diverse galectin-3 functions seem related to the ligands engaged in different cell types and tissues.
As a lectin, galectin-3 recognizes preferentially polylactosamines found in tri- or tetra-antennary N-linked oligosaccharides. Biosynthesis of these structures depends on the activity of N-acetylglucosaminyltransferase V (Mgat-5). Mgat-5 is responsible for the β1,6 branching of N-linked oligosaccharides and has been described as a transforming gene product (Demetriou et al. 1995; Yamamoto et al. 2000; Handerson and Pawelek 2003). The plant lectin leukoagglutinin (L-PHA) has been used for the identification of the Mgat-5-dependent glycosylation pattern (Dennis and Laferte 1989). Both L-PHA and galectin-3 have similar binding specificities (Demetriou et al. 2001). The former has been used as a probe for histochemical studies of human tumors from different origins (Sivridis and Agnantis 1996; Suzuki et al. 1999). To exploit the functional significance of this aberrant pattern of glycosylation, we have generated a novel probe based on human galectin-3 itself fused with the gene of the bacterial alkaline phosphatase (AP). We have therefore constructed a bifunctional probe, which has the binding activity of human galectin-3 and displays the enzymatic activity of AP, thus allowing for the in situ identification of galectin-3 ligands in tissue sections, in addition to its use in a variety of immunochemical methods.
Materials and Methods
Materials
All reagents were purchased from Sigma (St Louis, MO) unless stated otherwise and were of analytical grade or higher. Either biotin- or digoxigenin-conjugated lectins used were L-PHA, Galanthus nivalis agglutinin, and peanut agglutinin, all purchased from Roche (Mannheim, Germany). Antidigoxigenin antibodies were purchased from Roche. Laminin-1 was a kind gift from Dr. Vilma Martins, Ludwig Institute for Cancer Research, São Paulo, Brazil.
Cells and Antibodies
Cell lines used were the murine melanoma B16-F10 and two sarcoma cell lines (CCR2 and Σ12). Murine melanomas were cultured in RPMI 1640, supplemented with 10% fetal calf serum (Gibco; Grand Island, NY) in a humidified incubator equilibrated with 5% CO2 at 37C. CCR2 and Σ12 cells were cultured in DMEM supplemented with 10% fetal calf serum under the same conditions. Cells were harvested at subconfluence with trypsin and EDTA. To inhibit the processing of N-linked glycans, cells were treated with the α-mannosidase inhibitors 1-deoxymannojirimycin (dMN, 1 mM for 24 hr) or swainsonine (SW, 1 μg/ml for 48 hr).
The hybridoma-secreting rat anti-galectin-3 monoclonal antibody (TIB166, M3/38) was purchased from the American Tissue Culture Collection (ATCC; Manassas, VA) and was maintained in DMEM supplemented with 10% fetal calf serum under culture conditions. Supernatants were harvested, pooled, and tested for activity and specificity using a panel of cell extracts and the purified recombinant protein galectin-3. Rabbit anti-bacterial AP was purchased from Chemicon (Temecula, CA), anti-rat IgG horseradish peroxidase (HRP) conjugates were from KPL, Kirkegaard and Perry Laboratories (Gaithersburg, MD), and anti-rabbit IgG HRP conjugates were from Sigma.
In Vivo Experiments
Female Balb-c and C57bl/6 mice (6–8 weeks old) were used in the experiments. Animals were housed under controlled environmental conditions in 12-hr light cycles. Samples from normal murine tissues (e.g., thymus) were collected after approval by the University of São Paulo Medical School Ethics Committee for Research (both on animals and humans). When indicated, animals were inoculated with either the melanoma cell line (B16F10, isogenic to C57bl/6 mice) or the CCR2 sarcoma cell line (isogenic to Balb-c mice). Animals were sacrificed when tumors reached 0.5 cm3. Tumors were then excised and processed for histopathological analysis.
Construction of the Hybrid Galectin-3 Molecule
The human galectin-3 (gal3) gene was cloned in the pLIP6-GN vector, which encodes the AP gene, phoA. This vector presents unique restriction sites, SfiI and NotI, between codons coding for residues +6 and +7 of mature AP, thus allowing the periplasmic exportation of the fusion protein and correct processing of the signal peptide, after induction of the tac promoter with isopropyl 1-thio-β-D-galactopyranoside (IPTG). This facilitates disulfide bond formation, solubility, extraction, and purification of proteins (Uhlen and Moks 1990). In the empty pLIP6-GN vector, the phoA gene is out of frame and advantageously restored upon cloning of the foreign DNA insert, permitting a visual selection of blue colonies on BCIP agar plates (Gillet et al. 1992). In addition, this vector also has an ampicillin-resistance cassette (ampr). AP produced by this system presents two mutations resulting in a variant enzyme with enhanced enzymatic activity (Muller et al. 2001). The pLIP5-GN vector was used to obtain a positive AP control because its phoA gene is in-frame, expressing after induction the variant active enzyme alone.
The gal3 gene was PCR amplified from the pKK322-Gal3 clone using primer gal3NTSfi (5′-attagggcccagccggccgcagacaatttttcg-3′) and primer gal3CTNot (5′-gagaggcggccgctatcatggtatatga-3′), which added a SfiI and a NotI restriction site at the 5′ and 3′ of the PCR product, respectively. The amplification product was digested with SfiI and NotI restriction endonucleases and subsequently ligated into the pLIP6-GN vector previously cleaved with the same enzymes (see scheme in Figure 1A). The ligation product was used to transform E. coli DH5α, and the bacteria were seeded in TSB–agar plates (tryptone 20 g/L, yeast extract 5 g/L, NaCl 5 g/L, and K2HPO4 2.5 g/L, pH 7.5, 1.5% agar) containing 100 μg/ml ampicillin, 100 μM IPTG, and 40 mg/ml BCIP. The phosphate in the media inhibited the expression of the bacteria phoA gene, whereas the IPTG induced the vector tac promoter, permitting the expression of AP fusion proteins that hydrolyzed the BCIP substrate resulting in blue colonies. Ampicillin-resistant blue colonies were PCR-screened using the pLIP forward sequencing primer (5′-CAAAGCACTATTGCACTGGC-3′) and the reverse gal3CTNot primer used in the amplification of the gal3 gene. A positive clone was selected and confirmed after complete sequencing of the gal3 gene and upstream AP coding fragments. DNA sequencing was carried out using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems; Foster City, CA).
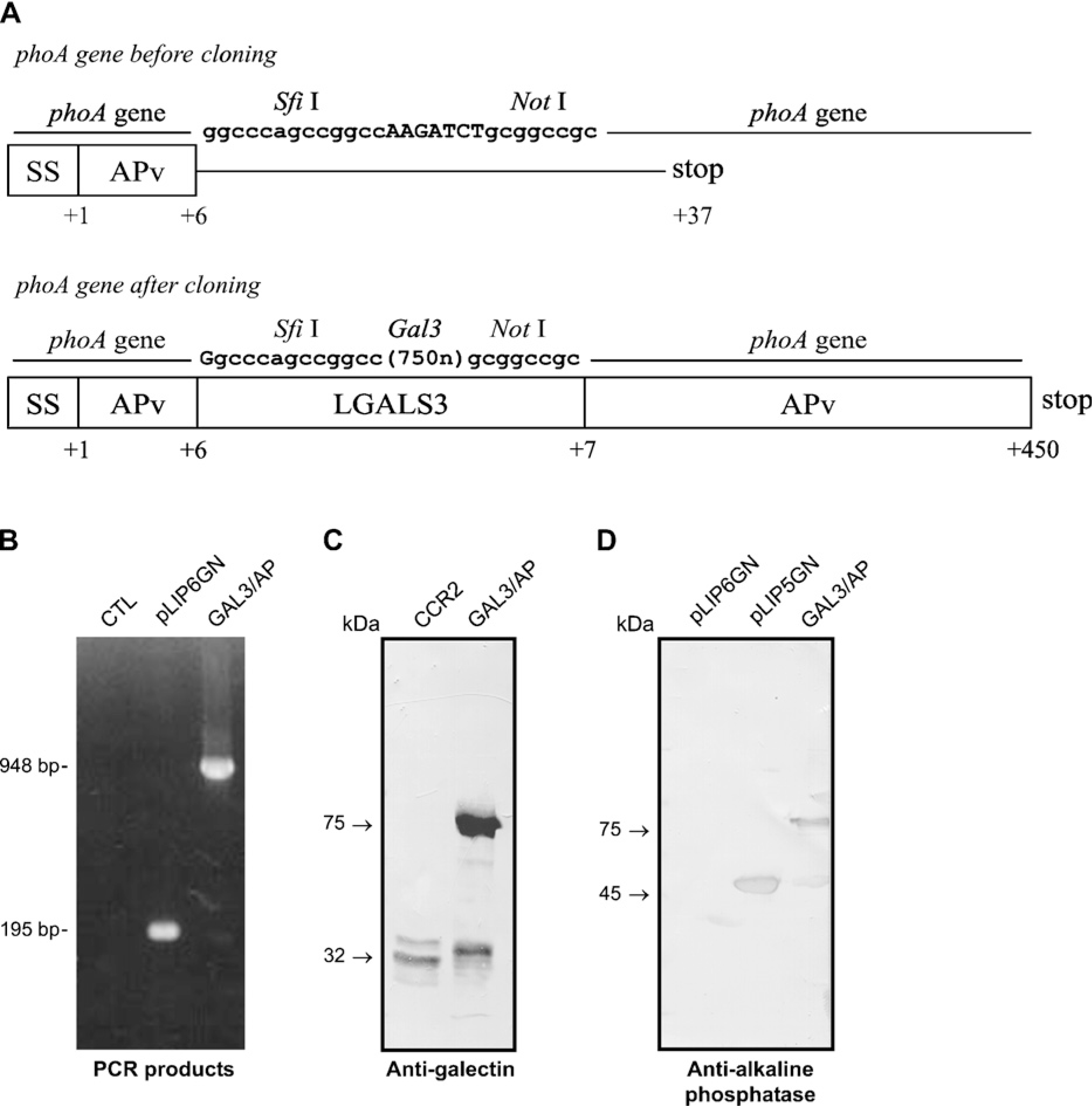
Construction and expression of a chimeric probe containing both galectin-3 and alkaline phosphatase (AP) activities. (
Protein Expression and Purification
A single clone was used to transform E. coli BL21 strain. The colonies were cultured in TSB medium supplemented with 100 μg/ml ampicillin at 37C until absorbance at 600 nm reached ∼0.6. The tac promoter was then induced with 100 μM IPTG for 4 hr at 37C. Bacteria from cultures were centrifuged, and the periplasm was extracted by cold osmotic shock as previously described (Ducancel et al. 1989).
Western and Lectin Blot Analysis
The presence of the recombinant fusion protein was detected by SDS-PAGE after staining with Coomassie blue or by Western blotting, using antibodies that recognize galectin-3 and AP or alternatively using the AP substrate, 5-bromo-4-chloro-3-indolyl phosphate in the presence of nitroblue tetrazolium solution (NBT/BCIP; Boehringer Mannheim, Indianapolis, IN). After periplasmic extraction, the samples were clarified by a 20-min centrifugation at 4C, and the protein concentration was determined using the BCA protein assay reagent (Pierce Biotechnology; Rockford, IL). Equal amounts of periplasmic extracts were boiled in SDS sample buffer under reducing conditions (240 mM Tris–HCl, pH 6.8, SDS 0.8%, glycerol 40%, β-mercaptoethanol 200 mM, and bromophenol blue 0.02%) for 5 min. Samples were separated by electrophoresis on 12.5% SDS–polyacrylamide gels under reducing conditions and transferred to a polyvinylidene difluoride membrane (Millipore; Bedford, MA). After protein transfer, the membranes were incubated with 5% non-fat dry milk dissolved in PBS (10 mM sodium phosphate, 150 mM NaCl, pH 7.2) and incubated with rat monoclonal anti-galectin-3 M3/38 or with 2 μg/ml rabbit polyclonal anti-bacterial AP overnight at 4C. After three washes with 5% non-fat milk in PBS, the membranes were incubated for 1 hr with 0.5 μg/ml HRP-conjugated rabbit anti-rat IgG or HRP-conjugated anti-rabbit IgG. Following three washes, the reaction was developed using 3.75 mg DAB in Tris-buffered saline (50 mM Tris–HCl, pH 7.5, containing 150 mM NaCl) in the presence of H2O2 (1 μl/ml; Fluka, Seelze, Germany).
For lectin blots, protein extracts were transferred to PVDF membranes, which were blocked with PBS-1% BSA and incubated with biotin-conjugated L-PHA (Phaseolus vulgaris agglutinin) or digoxigenin-labeled GNA (Galanthus nivalis agglutinin) in PBS containing 2% BSA overnight at 4C. Next, the membranes were washed with TBS (150 mM NaCl, 50 mM Tris–HCl, pH 7.5) and incubated with the appropriate AP-conjugated secondary reagents. Following three washes with TBS and one wash with TBS containing 1 mM MgCl2, 1 mM MnCl2, and 1 mM CaCl2, the membranes were developed using the AP substrate NBT/BCIP. Membranes were also incubated with 30 μg/ml gal3/AP in PBS containing 1% BSA overnight at 4C, washed with TBS, and developed as described above.
Enzyme-linked Lectin Assay (ELLA) and Statistical Analysis
ELLA was performed to evaluate the lectin and AP activity of gal3/AP. To assay the lectin activity, wells from a flat-bottom microtiter plate from Corning (Corning, NY) were coated with 10 μg/ml of laminin-1 for 1 hr at 37C and washed with PBS. Nonspecific sites were blocked with 1% BSA for 30 min at 37C and washed with PBS. Increasing concentrations of gal3/AP periplasmic extract (in the range of 0–20 μg/ml) were added to the wells either in the absence or presence of 100 mM of lactose (Sigma) and incubated overnight at 4C. After washing, because affinity constants of galectin-3 binding are in the micromolar range, the bound lectin was then cross-linked with 1% paraformaldehyde for 15 min. The wells were then washed three times with blocking solution and incubated with rat monoclonal anti-galectin-3 M3/38 for 1 hr at 37C. After three washes, 0.5 μg/ml rabbit anti-rat IgG HRP conjugate was added and incubated for 1 hr at 37C. Following washes, the freshly prepared substrate (0.4 mg/ml o-phenylenediamine in 1 ml of 0.05 M citrate–phosphate buffer pH 5.0, containing 10 ml H2O2 30%) was added to each well. The reaction was run protected from the light and stopped through addition of 3 N H2SO4. Absorbance was read at 492 nm in a multiscan EIA reader (Bio-Rad Laboratories; Hercules, CA). Alternatively, to assay the AP activity, after incubation with gal3/AP, p-nitrophenylphosphate (1 mg/ml in diethanolamine buffer) was added. The reaction was run protected from light and, after color developed, absorbance was read at 415 nm in a multiscan EIA reader. Statistical analysis was done using two-way ANOVA (GraphPad PRISM 4 software), and the comparisons were determined significant at the 5% level (p<0.05).
Direct Binding Assay
Direct binding assay was performed to evaluate the fine specificity of gal3/AP. Diverse glycans (Glycan Array version v2.3, NIGMS Consortium for functional glycomics) were added in a 384-well HBC NeutrAvidin Black plate (ligand coating, 30 pmol/well). For the complete list of glycans used in this assay, please consult the site http://www.functionalglycomics.org/static/consortium/resources/resourcecoreh2.shtml. The plate was then washed with PBS with 0.05% Tween 20 (wash buffer) and incubated with 15 μg/ml gal3/AP in wash buffer and 1% BSA. The bound lectin was detected with anti-AP antibodies followed by incubation with Alexa 488-conjugated secondary antibodies, using a plate fluorimeter.
Tissue Staining
Sequential tissue microarray slides containing panels of human tumors of diverse origins and their normal counterparts were obtained from the Hospital A. C. Camargo, São Paulo, Brazil after approval from the local Ethics Committee and tumor bank director. Sections of buffered paraformaldehyde-fixed, paraffin-embedded thymuses or experimental murine sarcomas or melanomas were also used. Tissue sections were routinely processed, and antigen retrieval was done by heating the specimens in citrate–phosphate buffer, pH 6.0, in the microwave for 10 min. After blocking with PBS-1% BSA, the sections were incubated with 30 μg/ml gal3/AP in the blocking solution overnight at 4C, either in the absence or presence of 100 mM lactose. After washing, the reaction was developed using Fast Red in Naftol phosphate Tris–HCl buffer (1 mg/ml; Dako, Carpinteria, CA). Specimens were also stained using anti-galectin-3, anti-CD34 antibodies, and L-PHA and then developed with peroxidase-conjugated secondary reagents.
Results
Production of the Gal3/AP Hybrid Protein
Galectin-3 cDNA was successfully subcloned into the pLIP6-GN vector, giving rise to the construct shown in Figure 1A. A blue colony was screened by PCR, which confirmed the presence of a 948-base pair band corresponding to the galectin-3 gene (Figure 1B). This clone was further sequenced confirming the correct in-frame cloning of the galectin-3 cDNA. The pLIP6/gal3 clone was subsequentially used to transform E. coli BL21. After induction, the periplasmic proteins were extracted by cold osmotic shock and the expressed chimeric form of galectin-3 (gal3/AP) was further analyzed by SDS-PAGE and confirmed by Western blotting analysis using antibodies that recognize galectin-3 (Figure 1C) and AP (Figure 1D). Protein extracts from CCR2 cells were used as a control; the polypeptide recognized by M3/38 had an apparent molecular mass of 32 kDa. In gal3/AP samples and under reducing conditions, M3/38 recognized bands with apparent molecular masses of 32 and 75 kDa. The 32-kDa band is likely galectin-3 derived from gal3/AP processing (Figure 1C). The anti-AP antibody also recognized two bands with apparent molecular masses of 45 and 75, under reducing conditions. The 45-kDa band is likely AP released from gal3/AP processing. The periplasmic proteins from pLIP6-GN and pLIP5-GN were used as negative and positive controls, respectively (Figure 1D). Under non-reducing conditions, a third band with apparent molecular mass of 175 kDa was also observed in Western blots using both anti-galectin-3 and anti-AP antibodies (data not shown). It is conceivable that dimers of gal3/AP could exist, explaining the 175-kDa band.
Gal3/AP Has Both Lectin and AP Activities
Periplasmic extracts were assayed for both AP and lectin activities. Galectin-3 interacts with polylactosamines containing glycoconjugates including laminin. To examine the lectin activity of gal3/AP, ELISA plates were coated with laminin-1, and increasing concentrations of gal3/AP were incubated in the presence or absence of 100 mM lactose. Figure 2A shows that gal3/AP bound to laminin-1 in a dose-dependent manner and that this interaction was inhibited by lactose. To investigate the AP activity of the chimeric protein, p-nitrophenylphosphate was used as substrate. As shown in Figure 2B, gal3/AP partially retains its phosphatase activity comparing to the product of pLIP5-GN, used as a positive control. In these experiments, the periplasmic extract of bacteria transformed with pLIP6-GN was used as negative control. The reduced phosphatase activity of the chimera may be attributed to the inclusion of the 316-amino acid residues of Gal-3 within the N-terminal portion of the AP molecule, impairing the optimal folding of the molecule.
Gal3/AP Has the Same Specificity of Parental Galectin-3
Direct binding of gal3/AP to a panel of >190 glycans (Consortium for Functional Glycomics, Glycan plate arrays, versions 1.2 and 2.7) allowed us to determine the specificity of the chimeric lectin. Table 1 indicates the eight distinct glycans recognized by gal3/AP with higher affinity. These are essentially the same glycans recognized by the parental molecule, using the same version of the glycan plate array (galectin-3, data from 2004, available at the site of the Consortium for Functional Glycomics, http://www.functionalglycomics.org, under CFG data, Core H-glycan screening). Signal-to-noise ratio is an indirect measurement of the apparent affinity. Sialylation did not interfere with gal3/AP binding (Table 1). This was further confirmed in lectin blots using sialidase-treated protein extracts (data not shown). Note that more recently the Consortium for Functional Glycomics extended the platform using a printed array, which now includes >250 different glycans. Among them, galectin-3 bound strongly to two other glycans that were not present in the plate array used herein, namely, Galβ1–4GalNAcaα1–3(Fucα1–2)Galβ1–4GlcNAcβ and GlcNAcβ1–3Galβ1–4GlcNAcβ1–3Galβ1–4GlcNAcβ which, however, share similar determinants to blood group A (type 2) and dilactosamine (LN)2, respectively.
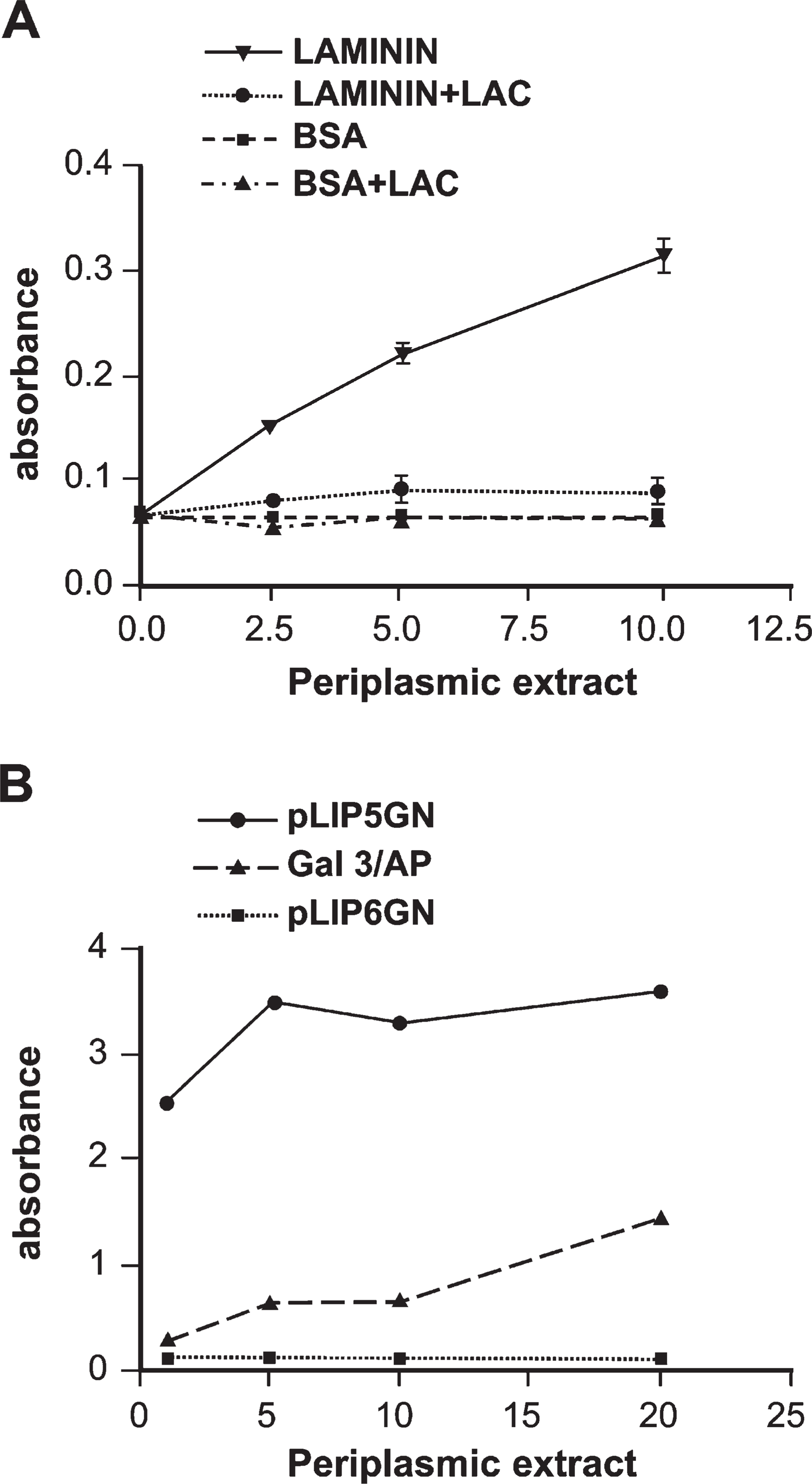
Gal3/AP shows both lectin and AP activities. (
Gal3/AP binding specificity
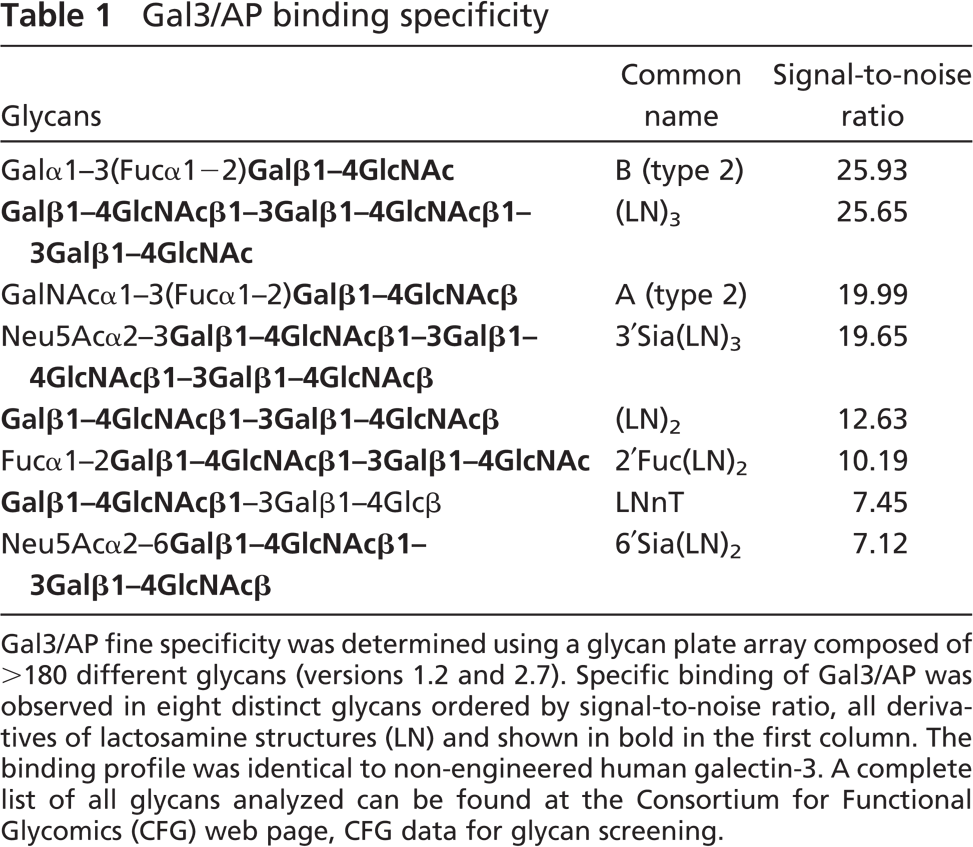
Gal3/AP fine specificity was determined using a glycan plate array composed of >180 different glycans (versions 1.2 and 2.7). Specific binding of Gal3/AP was observed in eight distinct glycans ordered by signal-to-noise ratio, all derivatives of lactosamine structures (LN) and shown in bold in the first column. The binding profile was identical to non-engineered human galectin-3. A complete list of all glycans analyzed can be found at the Consortium for Functional Glycomics (CFG) web page, CFG data for glycan screening.
Gal3/AP Recognition Depended at Least in Part on the N-linked Oligosaccharides of Different Glycoproteins
Gal3/AP was useful in ligand blottings, as shown in Figure 3. The pattern of gal3/AP binding was compared with L-PHA binding in protein extracts derived from established cell lines. To evaluate the binding of gal3/AP to N-linked oligosaccharides, the sarcoma cell lines CCR2 and Σ12 were treated with 1 mM of dMN, an inhibitor of α-mannosidase I. After the treatment, protein extracts were separated in 10% SDS–polyacrylamide gels under reducing conditions, transferred onto PVDF membrane, and analyzed with L-PHA, gal3/AP, and GNA. L-PHA recognizes β1–6-GlcNAc-bearing glyco-proteins and was used as positive control. GNA recognizes high-mannose N-linked oligosaccharides, which were exposed after dMN treatment and, therefore, were used as controls for the treatment with dMN. Two major glycoproteins of apparent molecular masses of 195 and 82 kDa were decreased after dMN treatment as revealed in L-PHA and gal3/AP lectin blot. In contrast, one major band of 160 kDa increased in GNA lectin blot (Figure 3A).
In a second set of experiments, B16F10 and Σ12 cells were treated with 1 μg/ml of swainsonine (SW), an inhibitor of mannosidase II, for 48 hr. After that, protein extracts from B16F10 and Σ12 cells were subjected to lectin blot analyses, using L-PHA and gal3/AP (Figure 3B). Several bands were recognized by L-PHA in B16F10 extracts. Major bands included a polydisperse band of 120–140 kDa and an 80-kDa band. The 120-to 140-kDa polydisperse band was also found in Σ12 cell extracts. This band was also recognized by Gal3/AP. Upon SW treatment, a significant decrease in the binding of both L-PHA and Gal3/AP was found. The 80-kDa band was no longer identified after SW treatment in B16F10 cells. On the other hand, two smaller bands of 50 and 60 kDa apparent molecular mass were observed upon SW treatment. It is clear from both approaches that L-PHA and gal3/AP binding may be similar but not identical and are at least in part dependent on N-linked glycosylation. Gal3/AP binding was abolished by co-incubation of the lectin with 100 mM lactose (data not shown), as observed in direct binding assays using laminin as a galectin-3 ligand (Figure 2A).

L-PHA and gal3/AP recognition patterns are similar but not identical. We compared the binding profiles of both leukoagglutinin from Phaseolus vulgaris (L-PHA) and gal3/AP using lectin blots of selected cell lines (the murine fibrosarcoma cell lines Σ12 and CCR2 and the murine melanoma cell line, B16F10). Cells were previously treated (+) or not (−) with the mannosidase inhibitors deoxymannojirimycin (DMN), an α-mannosidase I inhibitor (
Expression of Galectin-3 Ligands in Different Types of Tumors by Tissue Assay
Gal3/AP could be used to identify galectin-3 ligands in situ. Paraffin-embedded murine thymuses (see example in Figures 4A–4D) and sarcomas (derived from CCR2 murine cell line, Figures 4E–4H) were used to compare the binding pattern of the plant lectin L-PHA (Figures 4B and 4F) and gal3/AP (Figure 4C, 4D, 4G, and 4H). Gal3/AP binding was abolished by coincubation of the lectin with 100 mM lactose (data not shown and supplemental figures, SF1–SF9), as observed in direct binding assays using laminin as a galectin-3 ligand (Figure 2A). Presence of galectin-3 is also shown in Figure 4 (A, E).
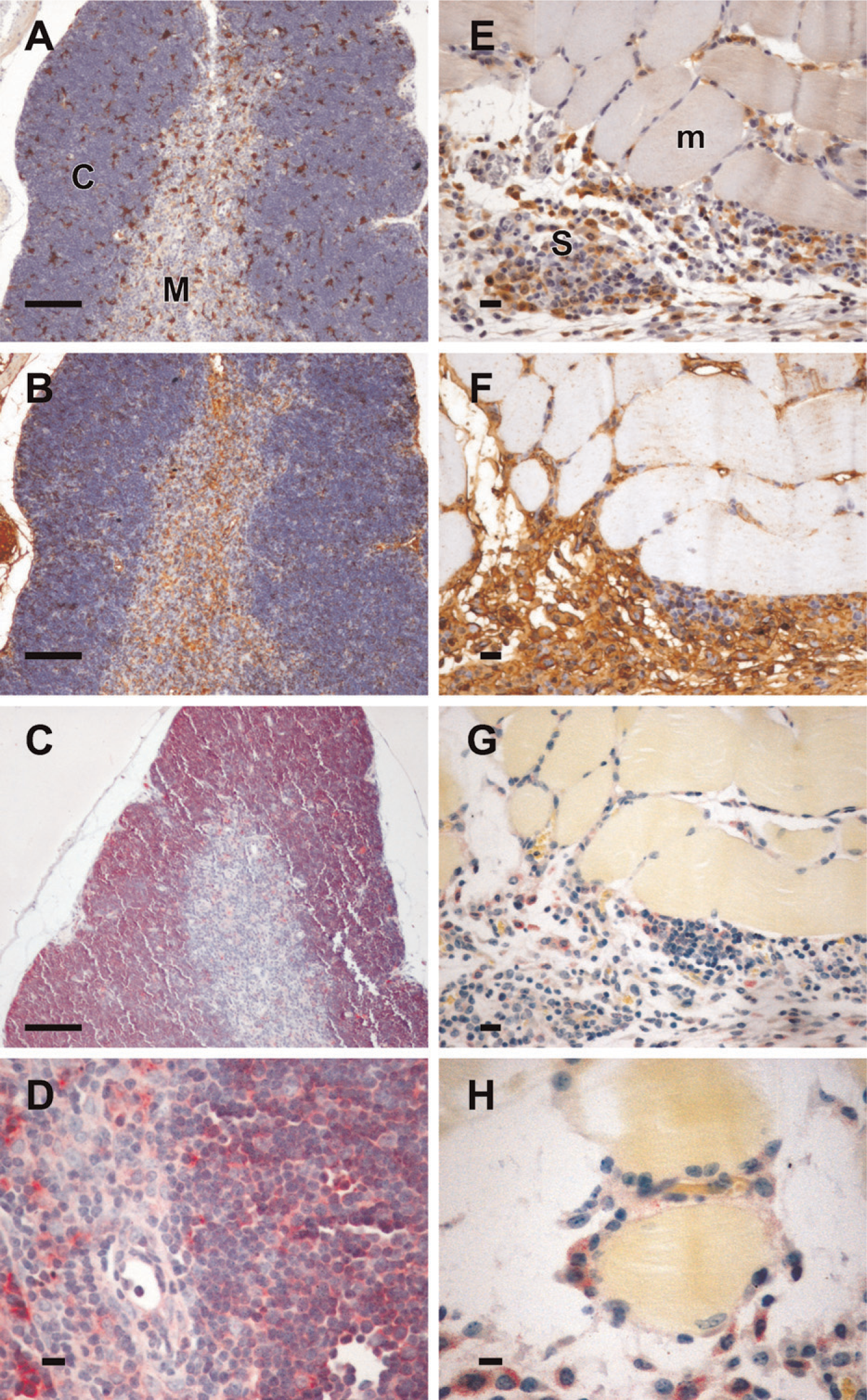
Lectin histochemistry using Gal3/AP indicates that not all L-PHA-positive cells bear galectin-3 ligands. Murine thymus (
In the murine thymus, cells expressing galectin-3 are scattered in cortex and medulla. Whereas L-PHA structures are found in both thymic compartments, especially in the vascular structures in the medulla, gal3/AP bound mainly cortical cells (Figure 4C) and a few cells in the medulla (Figure 4D). De novo expression of sialyltransferases is a common hallmark of thymocyte differentiation (Wu et al. 1997; Starr et al. 2003). As mentioned above, loss of galectin-3 binding was not due to masking of galectin-3 ligands by the addition of sialic acid residues because treatment of thymocyte extracts with sialidase did not expose novel gal3/AP binding sites (data not shown). Experimental sarcomas (Figures 4E–4H) express galectin-3 (Figure 4E) and migrate onto basement membranes of muscle fibers, as discussed elsewhere (Jasiulionis et al. 1996) and indicated in Figure 4H (note that Gal3/AP-stained sarcoma cells are adhered to muscle fibers). In this example, gal3/AP binding was not as evident as L-PHA binding, although as shown in Figures 4F–4H, infiltrating sarcoma cells stained with both plant and animal lectins.
Specimens from different human tumors were also analyzed using a tissue microarray. Examples of tissue staining by Gal-3/AP that were more representative are shown in Figure 5, which depicts examples of staining from breast carcinomas (Figures 5A and 5B), prostate carcinomas (Figures 5C and 5D), a glioblastoma (Figure 5E), and a melanoma (Figure 5F). Strong immunoreactivity was detected in the apical portion of well-differentiated breast carcinomas, including in the lumen secretion (Figure 5A). A trend to lose gal3/AP binding was observed in poorly differentiated tumors (Figure 5B). A similar trend was observed in prostate cancer (Figure 5C illustrates a well-differentiated tumor; Figure 5D illustrates a poorly differentiated tumor). Stromal reactivity was intense and found either in vascular elements (Figure 5D) or reactive glial cells (as indicated in the glioblastoma, arrow in Figure 5E). Heterogeneity of gal3/AP binding was observed in human melanomas, as illustrated in Figure 5F (arrow indicates an example of a strongly reactive cell in an infiltrated microenvironment). Supplemental figures illustrate the broad pattern of galectin-3 ligand distribution in a selected variety of human tumors, including squamous cell carcinoma, breast, prostate, pancreas carcinomas, melanomas, neuroendocrine tumors, neuroblastomas, and glioblastomas.
In murine melanomas (Figure 6), gal3/AP bound both vascular structures and infiltrating stromal cells. Gal3/AP binding (Figures 6B and 6D) was comparable to anti-CD34 antibodies, as illustrated in Figures 6A and 6C. In this regard, the probe we have developed could be used to determine microvascular density within experimental tumors. Moreover, a subset of dendritic-like stromal cells (Figure 6D) also stained intensely with gal3/AP.
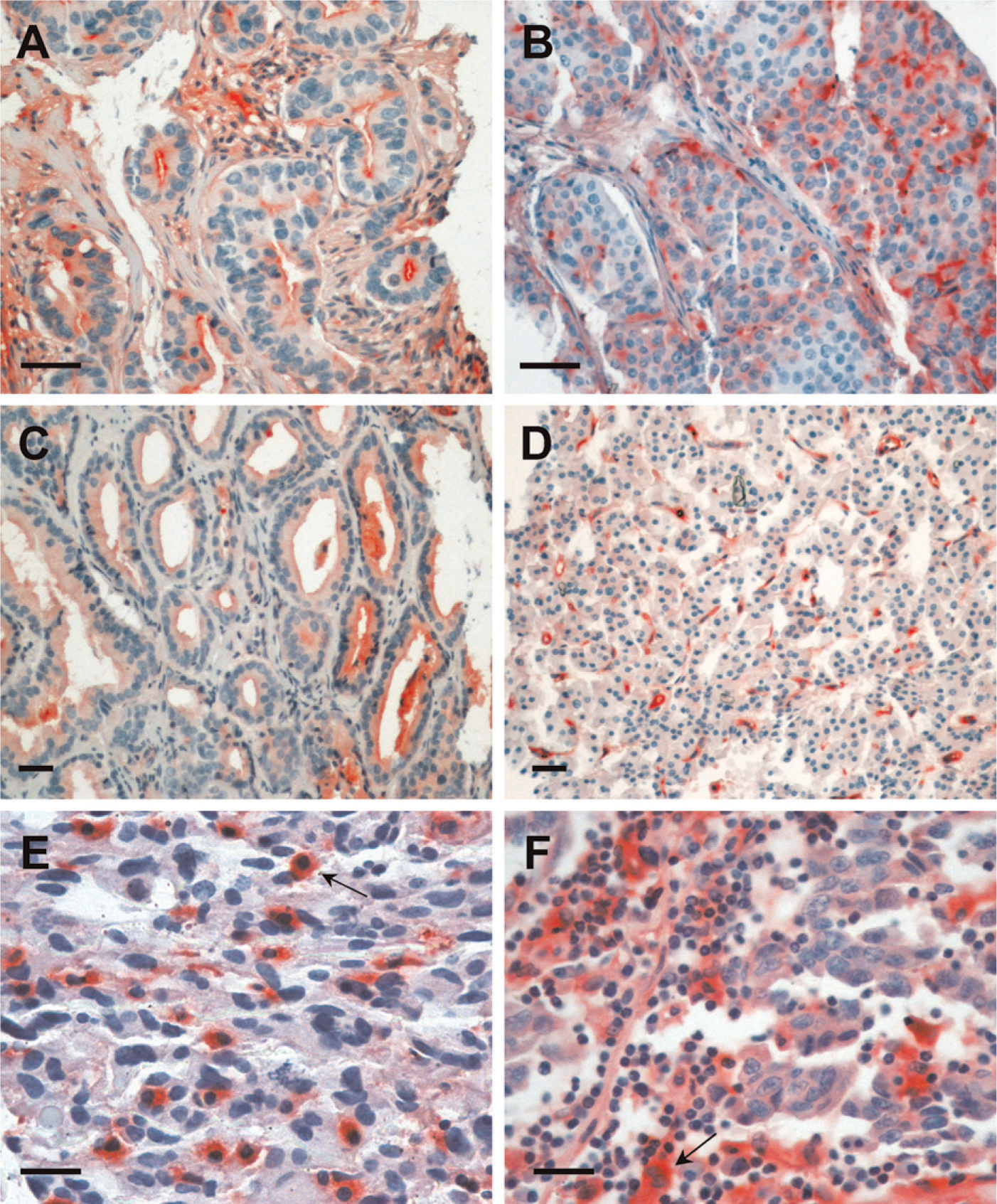
Human tumors are recognized by gal3/AP. Examples of the heterogeneous reactivity of gal3/AP in different human cancers. Breast (
Discussion
We have described here the production, expression, and some applications of gal3/AP, a hybrid molecule displaying both the carbohydrate binding properties of galectin-3 and enzymatic activity of AP. The binding profile of this chimeric molecule was compared with the binding of the plant lectin L-PHA. Although the binding specificities of L-PHA and galectin-3 are similar at the oligosaccharide level (Cummings and Kornfeld 1982; Sato and Hughes 1992; Demetriou et al. 2001), we showed here that neither glycoprotein profiling nor in situ binding of each lectin was identical. Therefore, it seems incorrect to use L-PHA and galectin-3 staining data interchangeably. As the probe presented here is derived from galectin-3 itself, it is a more adequate tool for pathophysiological studies on the roles of galectin-3 than the plant lectin. Gal3/AP will be useful to identify the binding patterns of galectin-3 and will be useful for the in situ determination of galectin-3 ligand distribution. This tool is an alternative to biotinylated lectins, which had been widely used to describe the distribution pattern of glycoconjugates in normalcy and malignancy. Indeed, biotinylated galectin-3 had been successfully used by different groups, following the pioneering work of Gabius and colleagues (Choufani et al. 1999; Plzak et al. 2001). Gal3/AP is an effective alternative to biotinylated galectin-3; the former, however, can be produced in a single step. As no chemical modifications are necessary for the generation of the chimeric probe, it is anticipated that Gal3/AP-specific activity would be rather constant from batch to batch, in addition to allowing a direct determination of galectin-3 ligands in situ, thus overcoming the potential problem of loss of specificity due to the use of secondary reagents for development of the reaction. Furthermore, Gal3/AP represents a suitable alternative for studying the distribution of galectin-3 ligands in tissues where endogenous biotin levels are high.
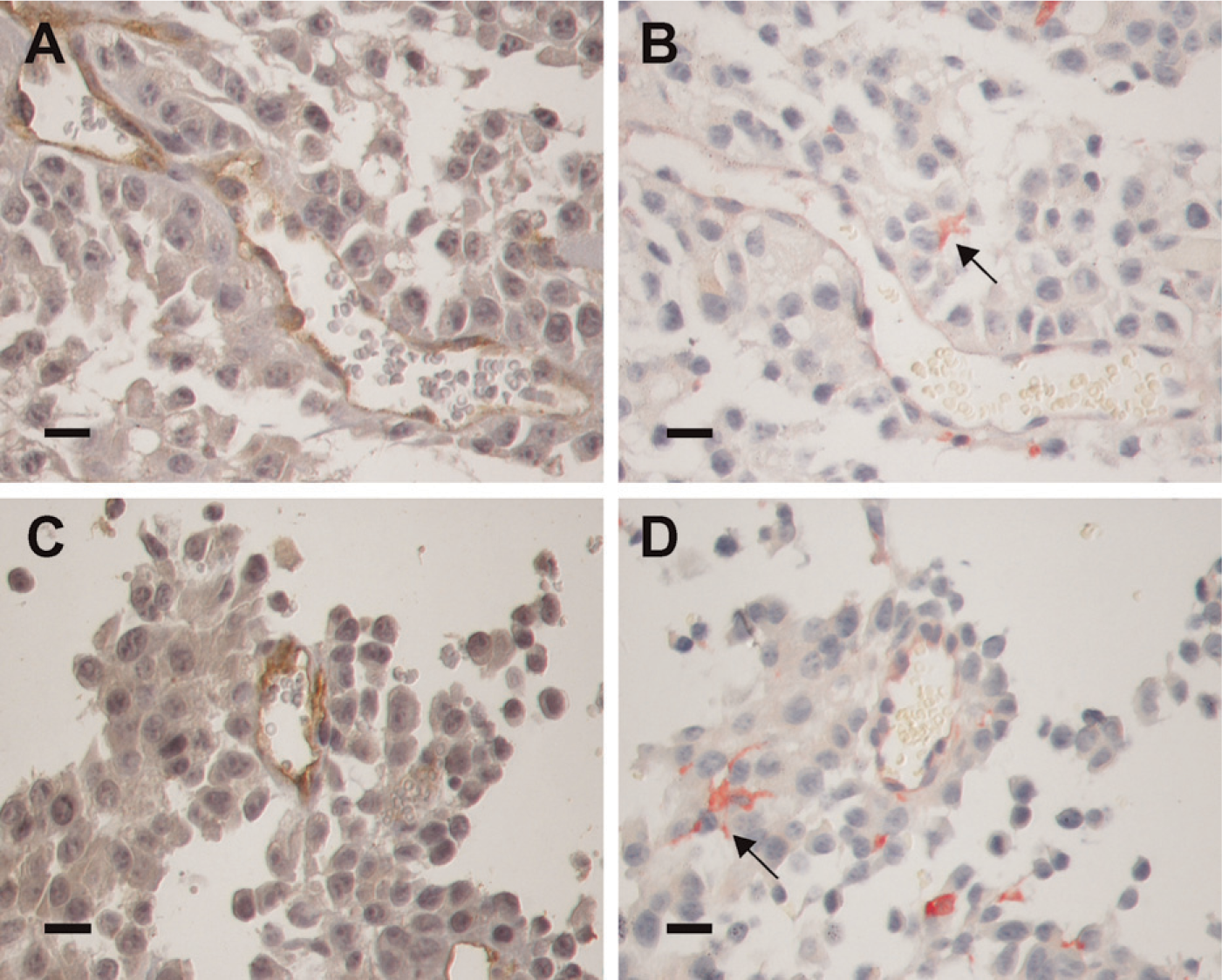
Gal3/AP is a useful probe for analysis of tumor microenvironmental cells. (
Immunopurified molecules can be probed directly with the chimeric galectin-3. Binding of the chimeric probe to immobilized glycoproteins in lectin blots has been used recently to identify different glycoforms of integrins as ligands for galectin-3 (Chammas and Bellis, unpublished data). The lectin blot and ligand overlay assays do not favor conformation-dependent protein–protein interactions, although there are a few examples of peptide–protein receptor interactions in ligand blottings in the literature (Chammas et al. 1994b). It seems likely that most of the binding partners of galectin-3 revealed in lectin blots are glycoproteins, and that the oligosaccharide residues present in the blotted glycoproteins are the actual determinants of galectin-3 binding. Indeed, lactose inhibited gal3/AP binding to blotted proteins (data not shown), as it did in ELISA assays (Figure 2A). Due to galectin-3 specificity, most proteins revealed in lectin blots are likely to be either cell surface or extracellular matrix glycoproteins. The finding that the mannosidase inhibitors, dMN and SW, led to a decreased binding of gal3/AP (Figure 3) corroborates the notion that N-linked glycans serve as galectin-3 ligands (Cheung et al. 1985; Partridge et al. 2004; Lagana et al. 2006). O-linked glycans, such as those present in CD43 and CD45, could also be recognized by galectin-3. On the other hand, intracellular glycoproteins do not usually present disaccharide structures, except in the case of cytokeratin, as described by Goletz et al. (1997).
There is increasing interest in determining the apparently non-glycosylated intracellular partners of galectin-3. Two hybrid systems led to the identification of molecules such as gemin 1 and 4 and β-catenin as galectin-3 binding molecules, among others (Park et al. 2001; Shimura et al. 2004). Optimal conditions for binding of gal3/AP to intracellular proteins may be determined in enzyme-linked lectin assays, a useful application of this chimeric molecule. These assays are likely to be done under reducing conditions, as those found in the cytoplasm.
The most direct application of the molecule described here is its use in direct lectin histochemistry. The involvement of galectin-3 in inflammation, development of both innate and adaptive immune responses, and tumor progression is anticipated by several lines of evidence and has been reinforced by studies using galectin-3 null mice (Colnot et al. 1998; Hsu et al. 2000; Bernardes et al. 2006; Silva-Monteiro et al. 2007). Although some of these roles may be exclusively due to the functions of intracellular galectin-3, some will involve its lectin function. The chimeric probe will aid in the identification and a more precise description of tissue microenvironments where galectin-3 may play a role.
In the murine thymus, for example, most of the ligands for galectin-3 are confined to the cortical thymocytes, different from what was observed to L-PHA staining. We have shown that extracellular galectin-3 acts as a de-adhesion molecule disrupting the interactions between thymocytes and thymic microenvironmental cells, such as the cortical thymic epithelial cells found in the thymic nurse complexes (Villa-Verde et al. 2002). Data shown in Figure 4D suggest that most of the galectin-3 ligands are in immature thymocytes found in the thymic cortex. This notion was recently confirmed by independent groups of experiments. On one hand, we have shown that galectin-3 mainly affects migration of CD4+CD8+ cells (Silva-Monteiro et al. 2007), whereas Baum's group demonstrated that galectin-3 induces CD4−CD8− cell death (Stillman et al. 2006). In experimental sarcomas, the probe was useful to illustrate the pattern of dysfunctional migration of sarcoma cells on laminin-rich muscle basement membranes (Figures 4E–4H).
However, the higher impact of the use of this probe will be on studies on the clinical significance of a specific pattern of aberrant glycosylation, namely, β1–6 branching of N-linked glycans. Although it is quite clear that several tumors accumulate glycoproteins bearing β1–6-branched N-linked oligosaccharides, the precise function of this altered pattern of glycosylation in glycoprotein function remains elusive (Dennis and Laferte 1989; Fernandes et al. 1991; Chammas et al. 1994a; Handerson and Pawelek 2003; Handerson et al. 2005; Siddiqui et al. 2005). An extremely interesting hypothesis on the role of this glycosylation pattern was revisited by Dennis (Partridge et al. 2004), based on a model proposed by Childs and Feizi, among others, in the 1980s. In this model, glycoproteins would exist as lattices on the cell surface. Maintenance of these lattices would depend on extracellular glycan-binding proteins such as galectin-3. Complexes of glycoproteins and lectins would render cells more sensitive to growth factors, whose interaction with their cognate receptors usually lead to receptor dimerization/oligomerization. If this is correct, expression of galectin-3 and its ligands may predict the prognosis of tumors. So far, most of the information on the presence of putative ligands for galectin-3 is based on the tumor reactivity to L-PHA. Determination of galectin-3 ligands using gal3/AP may represent a more suitable approach to this problem. As we have shown here, gal3/AP was useful for staining tissue mi-croarrays. Staining of specific tumor collections such as breast and prostate (see examples in Figure 5) and the correlation of the data obtained with clinical information will be useful to determine the significance of the presence of ligands for galectin-3 in tumor cells. Gal3/AP staining evidenced the loss of polarity in breast and prostate carcinomas. This event is a frequent finding in carcinoma progression and can be followed by the presence of tumor-associated antigens in the sera of patients. Examples for these antigens include secretion of CEA, which is a galectin-3 ligand (Cvejic et al. 2000; Wojciechowicz et al. 2000), and several sialomucins (such as CA125 and CA19.9), which may also bear polylactosamines recognizable by galectin-3. Gal3/AP will also be useful for the detection of aberrantly glycosylated glycoproteins in the sera of patients, using quantitative AP-based assays.
The survey in different tumors clearly indicated that not only tumor cells, but also microenvironmental cells as infiltrating leukocytes, express galectin-3 ligands (Figure 5 and Figure 6). Positive identification of these ligands is warranted, and gal3/AP will be a useful tool for this objective. Moreover, gal-3/AP stained vascular structures in tumor sections as well as the commonly used antibodies anti-CD31 and anti-CD34 (Figure 6). Accordingly, recent findings indicate that galectin-3 participates in tumor angiogenesis (Nangia-Makker et al. 2000; Fukushi et al. 2004; Shekhar et al. 2004) This finding will be useful to determine the microvascular density of tumors that do not express CD34 or in tumors that aberrantly express CD34, as is the case for experimental renal carcinomas (data not shown).
Finally, as Gal3/AP is a recombinant chimeric probe, its production in E. coli renders it a cost-effective tool for a variety of enzymatic assays including in situ identification of galectin-3 ligands and will help to determine the biological functions of this animal lectin.
Footnotes
Acknowledgements
This work was funded by FAPESP (Center for Cell-based Therapy Research, 1998/14247-6) and CNPq grants. The glycan-array analysis was conducted by the Protein–Carbohydrate Interaction Core (H) of The Consortium for Functional Glycomics funded by the National Institute of General Medical Sciences (Grant GM62116).
We are grateful to Dr. Frederic Ducancel, CEA-Saclay, Gif-sur-Yvette, France, for providing the vectors used in this work and to Dr. Paulo Lee Ho, Centro de Biotecnologia do Instituto Butantan, for sequencing the constructs. We thank the Core H staff, Ms. Angela Lee, and Mrs. Carole Davis for their help in conducting the assays.