
Abstract
Retinoic acid receptor β (RARβ) has been proposed to act as a tumor suppressor in breast cancer. In contrast, recent data have shown that RARβ promotes ERBB2-induced mammary gland tumorigenesis through remodeling of the stromal compartment and activation of cancer-associated fibroblasts. However, it is currently unknown whether RARβ oncogenic activity is specific to ERBB2-induced tumors, or whether it influences the initiation and progression of other breast cancer subtypes. Accordingly, we set out to investigate the involvement of RARβ in basal-like breast cancer using mouse mammary tumor virus (MMTV)-wingless-related integration site 1 (Wnt1)-induced mammary gland tumorigenesis as a model system. We found that compared with wild type mice, inactivation of Rarb resulted in a lengthy delay in Wnt1-induced mammary gland tumorigenesis and in a significantly slower tumor growth rate. Ablation of Rarb altered the composition of the stroma, repressed the activation of cancer-associated fibroblasts, and reduced the recruitment of inflammatory cells and angiogenesis. Reduced expression of IGF-1 and activity of its downstream signaling pathway contribute to attenuate EMT in the Rarb-null tumors. Our results show that, in the absence of retinoid signaling via RARβ, reduced IGF-1 signaling results in suppression of epithelial-mesenchymal transition and delays tumorigenesis induced by the Wnt1 oncogene. Accordingly, our work reinforces the concept that antagonizing RARβ-dependent retinoid signaling could provide a therapeutic avenue to treat poor outcome breast cancers.
Introduction
Retinoic acid (RA), which is derived from dietary vitamin A, plays an essential role in the maintenance of the differentiated state of epithelial cells and tissue remodeling [1–3]. The family of retinoic acid receptors (RARs) that includes the RARα, β and γ subtypes and associated alternatively spliced isoforms are the main mediators for the biologic effects of RA [4, 5]. RA and other natural and synthetic RAR agonists are well known to inhibit the growth of human breast tumor cell lines in vitro and in mouse xenograft models, a process that generally involves the induction of both apoptosis and cell-cycle arrest [6–12]. In addition, the absence of RARβ-dependent retinoid signaling has been associated with the development of carcinomas, including breast cancer, and resistance to the anti-cancer activity of RA [13]. Despite the potent growth-suppressing activity of retinoids on human breast cancer cell lines, this class of compounds has not  shown relevant anti-cancer activity in the treatment of breast cancer [14, 15].
shown relevant anti-cancer activity in the treatment of breast cancer [14, 15].
Recently, RAR-null mice have been used to address the role of the RARs in mammary gland development and the response of the gland to specific oncogenic insults [16, 17]. Ablation of RARα1 led to an increase in the density of the mammary epithelial tree and the content of luminal mammary progenitors, in addition to reducing the size of the mammary stem cell-containing compartment [17]. In contrast, loss of RARβ resulted in a delay in mammary tree development in the pubertal stage as a significant shortening of the distance between the lymph node and the terminal end buds were observed in the RARβ-null gland [16]. The developmental abnormalities observed in both RAR-deficient mouse models were transient since, as previously reported [18–20], the glands were fully functional at later stages of development. While ablation of RARα1 and β resulted in distinct developmental mammary gland abnormalities, the absence of RA signaling transduced by either receptor resulted in a delay in mammary gland tumorigenesis. In the RARα1-null model, the reduction observed in the mammary stem cell-containing compartment might be a factor in the delay in tumor formation by the Wnt1 oncogene, but the exact mechanism responsible for the prooncogenic property of RARα1 in this context remains to be determined. On the other hand, absence of RARβ led to a remodeling of the stroma during tumor progression induced by the Erbb2 oncogene that was marked by a decrease in angiogenesis, in the recruitment of inflammatory cells and in the number of myofibroblasts [16]. In agreement with this observation, tissue recombination experiments demonstrated that the presence of RARβ in the stromal compartment is essential for the growth of mammary carcinoma, and that RA-dependent activity of the Cxcl12/Cxcr4/Erbb2 signaling axis played a significant role in this process. Together, these studies suggest that retinoid-based approaches for the prevention and treatment of breast cancer should be re-evaluated and that a better understanding of the mechanisms of retinoid action in mammary gland tumorigenesis is required to achieve this goal.
Estrogen receptor β (ERβ) is an attractive target for drug development. While orginal models of estrogen action suggested that only a single ER gene (which encodes a protein that is now termed ERα) was responsible for transducing signals of estradiol and other ligands [11], the discovery in 1996 of a second ER gene, encoding ERβ, prompted a reevaluation of the estrogen signaling system. It is now known that ERα and ERβ play different roles in gene regulation [12] and that ERα and ERβ have overlapping but distinctive tissue distributions and non-redundant roles [13]. These considerations have led to the suggestion that ERβ could be an attractive therapeutic target for the development of selective agonists to treat and prevent neurodegenerative diseases [14] and other diseases, including autoimmune diseases, endometriosis, depression, hypertension, and colon, breast, prostate, lung, and skin cancer [15]. It is important that such ligands should not cross-react with ERα, which triggers classical estrogenic side effects, such as breast or uterine stimulation, thereby increasing a woman's chance of developing breast or uterine cancer, and gynecomastia and decreased libido in men [16]. Presently, natural and synthetic estrogens for ERβ are being studied in colon cancer, breast cancer, lung cancer, schizophrenia, and metabolic syndrome [17]. While the agonist ERB-041 failed to demonstrate efficacy in a Phase II double-blind clincial trial for rheumatoid arthritis, further studies are warranted to examine ERβ-selective efficacy in other inflammatory disorders [18]. With many other clinical trials still in progress awaiting completion of the study followed by publication of the findings, it is still too early to make any definitive conclusions about these drugs and their effects. The discovery of potentially beneficial effects of selective ERβ ligands on prostate cancer proliferation and apoptosis in the absence of full ERα or ERβ agonism has raised hopes that applications for new safe selective ERβ modulator ligands could emerge in the context of this disease [19–22].
In the current study, we further tested the specificity of the RAR/oncogene interaction in the development of mammary gland carcinoma using bigenic mice generated by crossing the RARβ-null mice with the MMTV-Wnt1 transgenic mice. The Wnt1 oncogene targets mammary stem cells or early progenitor cells as transformation cells [21]. Accordingly, Wnt1-induced tumors show a co-existence of both luminal cells and basal/myoepithelial cells, which is reminiscent of human basal-like breast cancer that is associated with poor prognosis and lack of effective treatment [22]. Wnt1-induced tumors have also been reported to have abundant activated stroma that correlated with more aggressive tumor development [23]. Therefore, we sought to investigate the influence of RARβ on Wnt1-induced mammary carcinoma. We found that inactivation of RARβ resulted in a protective effect against Wnt1-induced mammary gland tumorigenesis, abrogation of Wnt signaling in both the epithelial and stromal compartments, and suppression of epithelial-mesenchymal transition (EMT) in the tumors. Our work indicates therefore that RARβ is essential for the bi-directional interaction between tumor and stromal cells and that RARβ specific antagonists may represent a novel therapeutic strategy to treat poor outcome breast cancer.
Materials and Methods
Mice
All animals were FVB/NJ background and housed in a pathogen-free facility at McGill University and were given a standard rodent diet and water ad libitum. MMTV-Wnt1 mice were purchased from the Jackson Laboratory [24]. To determine the initiation of mammary tumor formation, mice were palpated weekly (sensitive to 1-mm tumor masses). All mouse manipulations were performed in accordance with the McGill Facility Animal Care Committee and the Canadian Council on Animal Care.
Cell culture
Mouse mammary fibroblast cell lines were generated from the MMTV-Wnt1 animals as described before [16]. To generate tumor cells, tumors were dissected, mechanically dissociated, and forced through 40 μm mesh. Viable cells were plated, grown in DMEM supplemented with 10% FBS and penicillin/streptomycin. The cells had virtually identical epithelial morphology under microscope and had similar cell surface marker expression. All cultured cells were maintained in DMEM supplemented with 10% FBS and incubated in 5% CO2 at 37oC. The fibroblasts were treated with all-trans RA (Sigma #R2625) at a concentration of 0.5 μM for 2 days to generate conditioned media. Conditioned media from Rarb-null or wild-type mouse mammary gland fibroblasts were also supplemented with either IGF-1 or anti-IGF-1 antibody (R&D Systems) at final concentrations of 10 ng/ml and 40 μg/ml, respectively.
Reactive stroma index
Hematoxylin and eosin staining was carried out in the histology service core of the Goodman Cancer Research Centre. Stained sections were examined in a blinded manner and scored for the reactive stroma index based on the percentage contribution of a stroma area in tumor mass (i.e., 0–10% stroma area = 0, 11–20% stroma area = 1, and so on, >50% stroma area = 5). The average of five areas was used as the reactive stroma index for each sample.
Histological analyses
Immunohistochemical analyses were performed on 4-μm formalin-fixed paraffin-embedded sections of tumors from mice killed one month after tumor initiation. Immunostaining was done using the VECTASTAIN avidin-biotin complex kit (Vector Laboratories) as previously described [46]. Immunofluorescence staining was conducted on formalin-fixed paraffin-embedded tumor sections. Five random areas per section from 3 separate sections obtained from different mice for each group were analyzed. The following dilutions of primary antibodies were used: rabbit polyclonal anti–human-RARβ (Abcam), 1:100; mouse monoclonal anti-human Ki67 (clone B56; BD Pharmingen), 1:100; rabbit polyclonal anti-mouse Wnt1 (Upstate), 1:50; rabbit polyclonal anti–human-caspase-3 (Cell Signaling), 1:50; rat monoclonal anti–mouse-CD31 (clone Mec13.3; Biocare), 1:50; mouse monoclonal anti-CD45, 1:100, and rabbit polyclonal anti-mouse collagen I, 1:100 (Abcam Inc.), horse polyclonal anti-human smooth muscle actin-α (α-SMA) (Abcam), 1:100,; rabbit polyclonal anti-mouse IGF1 (Abcam), 1:100. For the negative control, primary antibody was replaced with non-immunized rabbit or mouse IgG (Vector Laboratories). All images were taken with 10x objectives (100x magnification). For multiple antigen labeling the VECTASTAIN System (Vector ABCDAB kit, Vector ABC-AP kit, and Vector ImmPRESSNovaRED and VIP kit) (Vector Laboratories) was used. Peroxidase and alkaline phosphatase substrates were used to develop color of the antigens with different localization. To compare positive cell numbers, 10 random fields per section were documented in the Aperio Image System and were analyzed using the Spectrum software (Aperio Technologies). Mean values shown beside the corresponding images were determined from results from at least six different mice for each genotype.
Apoptosis analysis
The number of TUNEL-positive or caspase 3-postive tumor cell nuclei was calculated relative to the total number of 1,000 cancer cell nuclei, which was analyzed with the Spectrum software. Mean values were determined from results from at least six different mice.
Western blotting
Lysates were prepared from the mammary tumors of three Rarb-/- and three wild-type mice. Antibodies against phospho-GSK3β (Ser9, 07-835), GSK3β (07-1413), Snail (ABD38), phospho-AKT (9611), and AKT (9272) were purchased from Cell Signaling Technology. Antibodies against RARβ (SC-552), Wnt-1 (SC-5630), Wisp1 (SC-13316), E-cadherin (SC-7870), integrin α5 (SC-10719), LAMA1 (SC-56145), vimentin (SC-373717), cytokeratin (SC-529), IGF1 (SC-7144), IGFBP5 (SC-6006), and actin (SC-1616) were purchased from Santa Cruz Biotechnology.
Laser capture microdissection, RNA extraction and linear amplification
All tissues included in this study were re-examined by an animal pathologist dedicated to the project. Tissue specimens were micro-dissected into epithelium and stroma using a PixCell IIe laser micro-dissection (LCM) system (Arcturus). All micro-dissections were performed within two hours following tissue staining. Total RNA was extracted from each population of micro-dissected cells using a guanidinium isothiocyanate (GITC) extraction protocol. Briefly, LCM caps were incubated for 5 minutes (room temperature) in 200 μl GITC extraction buffer (4 M GITC, 25 mM sodium citrate pH 7.0, 0.1 M β-mercaptoethanol, 0.5% N-lauroylsarcosine) supplemented with 1.6 μl β-mercaptoethanol. Subsequently, 20 μl of 2 M NaOAc, pH 4.0, 220 μl of water-saturated phenol and 60 μl of chloroformisoamyl alcohol (23:1) were added to the extraction buffer. Following a 15 minute incubation on ice and centrifugation (12,000 rpm, 15 minutes) the aqueous phase was removed and RNA was precipitated with 2 μl glycogen (GenHunter, Nashville, Tennessee, USA) and 200 μl isopropanol. Samples were placed at −80°C for 30 minutes and centrifuged at 4 °C (12,000 rpm) for 30 minutes to pellet RNA. Pellets were washed with 70% ethanol, air-dried and subjected to DNase I treatment (Roche, Basel, Switzerland). DNAse treatment was performed in the presence of an RNase inhibitor (Invitrogen, Carlsbad, California, USA). Subsequently, samples were re-extracted as described above and re-suspended in 10 μl of diethylpyrocarbonate-treated water. RNA was quantified using a RiboGreen assay (Molecular Probes, Carlsbad, California, USA). Subsequently, 2 to 4 ng of total RNA was subjected to two rounds of T7 linear amplification using Ambion Amino Allyl MessageAmp kit (Ambion, Austin, Texas, USA) and labeled with Cy3 and Cy5 dyes according to the manufacturer's procedure. Prior to microarray hybridizations, amplified products were quantified using a spectrophotometer (Nanodrop, Wilmington, Delaware, USA) and analyzed using a BioAnalyzer to assay for quality (Agilent Technologies, Santa Clara, California, USA).
RNA microarrays
SurePrint G3 mouse Genome 8×60 K arrays (Agilent Technologies, product G4852A) were used for all experiments. RNA samples (500 ng) were subjected to fragmentation followed by 18 hours hybridization, washing, and scanning (Agilent Technologies, model G2505B) according to the manufacturer's protocol (manual ID #G4140-90030). Samples were hybridized against Universal mouse Reference RNA (Stratagene, ID #750600, La Jolla, California, USA). Duplicate hybridizations were performed for all samples using reverse-dye labeling. Microarray data were feature extracted using Feature Extraction Software (v. 7.11) from Agilent with the default parameters. Raw data were uploaded to the NCBI Gene Expression Omnibus database (GSE56391). Outlier features on arrays were flagged by the software. Arrays were required to have an average raw signal intensity of 1,000 in each channel, and a signal to noise ratio above 16 per channel. MvA plots were examined for signs of hybridization or labeling problems. Replicate arrays were required to have a concordance above 0.944. This level was established empirically using sets of known good replicate arrays in our database.
Real-time PCR for Igf1 expression
Total RNA from LCM samples was amplified using Amino Allyl MessageAmp II aRNA Kit (Ambion #1753). cDNA was made from 2 μg of RNA by reverse transcription with Oligo(dT) primer, dNTPs, 5X 1st strand buffer, DTT, RNase inhibitor, and Superscript II RNase H Reverse Transcriptase (Invitrogen). cDNA was amplified by qRT-PCR using the specific forward 5'-TTCTACCTGGCGCTCTGCTTGC and reverse 5'-CCCTCCGAATGCTGGAGCCATA primers, a QuantiTect SYBR Green PCR Kit (Qiagen) and a LightCycler instrument (Roche) following the Qiagen software protocol.
ELISA
Submandibular venous blood was collected into microcentrifuge tubes using a sterile lancet (Medipoint, Mineola, NY) according to the manufacturer's instructions. Whole blood was centrifuged at 2500xg for 20 minutes at 4 °C to coll ect serum, which was then stored at −80°C. Serum was processed for ELISA of IGF-1 (R&D Systems, Minneapolis, MN) according to the manufacturer's protocol. Serum IGF-1 levels are reported in ng/ml. An IGF-1 control sample, with aliquots stored at −80°C, was included on each plate, and all data are reported using simple ratio normalization to the initial reading of the control sample.
Statistical analysis
Two-tailed paired Student t test was calculated by Excel software. Prism software was used for one or 2-way ANOVA and log-rank (Mantel-Cox) test.
Results
Global ablation of Rarb in mice delays Wnt1-induced mammary gland tumorigenesis and alters the stromal compartment
To further study the potential role of RARβ as a tumor promoter in mammary gland tumorigenesis, we used the well-characterized mouse model of human breast cancer expressing the oncogene Wnt1 in combination with the Rarb-null allele [18, 24]. While both Rarb-/-and wild-type mice displayed similar high penetrance for tumor formation (78% and 83%, respectively), the Rarb-/- mice showed a significant delay (∼12 weeks, P<0.01) in tumor formation when strong expression of Wnt1 was driven by the MMTV promoter (Figure 1A). In addition, the tumors in mice with the Rarb-/- genetic background grew at a significantly slower rate than tumors induced in their wild-type siblings (6.3 vs 4.0 weeks, P<0.05) (Figure 1B). Consistent with these results, examination of mammary gland sections taken from MMTV-Wnt1 mice by stroma index than tumors developing in Rarb-null siblings (3.8±0.7 vs 2.9±1.1 P<0.05) (Figure 1F). To determine whether the absence of RARβ affected the growth potential of cancer associated fibroblasts (CAFs), we generated stromal fibroblasts from control and Rarb-/- mammary glands as previously described [16] and compared their growth rate in vitro. In low serum conditions (2% FBS), the Rarb-/- fibroblasts displayed growth arrest whereas wild type fibroblasts survived and divided indicating that Rarb-/- CAFs have less aggressive malignant properties that persists ex vivo (Figure 1G).

Ablation of Rarb suppresses Wnt1-induced mammary tumorigenesis and affects the composition of the stroma.
Loss of Rarb results in remodeling of the stromal compartment in Wnt1-induced mammary gland tumors
Stroma may initiate a desmoplastic reaction that includes activation of CAFs and trans-differentiation of tumor or epithelial cells into myofibroblasts, infiltration of immune cells, increased secretion of growth factors and cytokines as well as elevated matrix synthesis and remodeling that manifests as matrix stiffening. Analysis of differential gene expression in the stroma and epithelial compartments using laser capture microdissection (LCM) showed that several genes associated with activation of fibroblasts such as Col3a1, Col5a1, Col5a3, Col6a2, Col6a3, Fn1, Fap and Vim are down-regulated in the stromal compartment of MMTV-Wnt1/Rarb-/- animals compared to their wild type siblings (Figure 2A). Morphologically, IHC analysis indicated reduced collagen deposition in MMTV-Wnt1/Rarb-/- tumor sections (Figure 2B), indicating decreased matrix stiffening. Moreover, significantly decreased smooth muscle actin (α-SMA) positive cells were found in MMTV-Wnt1/Rarb-/- tumor sections (Figure 2C), further indicating that fibroblast activation was suppressed in these animals. In addition, a reduction in the recruitment of inflammatory cells as measured by CD45 staining was observed in the peritumoral stroma of Rarb-null mice relative to their wild type siblings (Figure 2D). LCM-RNA microarray data also showed that several genes involved in angiogenesis, including Angpt2, Angptl4 and Bmper, were down regulated in the stroma of MMTV-Wnt1/Rarb-/- animals (Figure 2A). Consistent with the gene expression data, non-uniformly distributed blood vessels with irregular shape were observed in sections obtained from the mammary tumors of wild type mice and stained for CD31 expression (Figure 2E). These abnormal blood vessels were inappropriately branched, dilated and usually ended blindly. In contrast, we found significantly fewer blood vessels in the Rarb-null tumors.
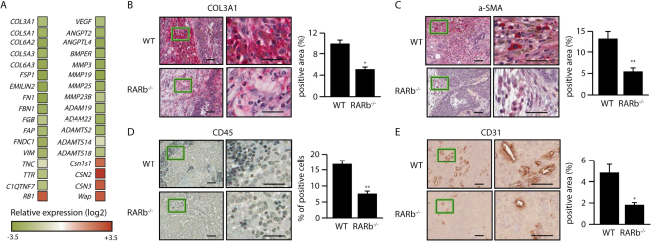
Loss of Rarb alters the gene expression profile of the stromal compartment and its composition.
Inhibition of epithelial-mesenchymal transition in Wnt1-induced Rarb-null tumors
Besides activation from local fibroblasts, emerging evidence indicates that matrix-producing myofibroblasts also arise through epithelial-mesenchymal transition (EMT) [25]. Interestingly, LCM-mRNA microarray data showed that several genes involved in EMT, such as Ecm2, Mmp3, Fgf2, and Igf1, were significantly down regulated in MMTV-Wnt1/Rarb-/- tumors (Figure 3A). We observed that the expression of markers of epithelium undergoing EMT, including E-cadherin, cytokeratin and laminin, were significantly increased at the protein level, whereas the expression of vimentin and integrin (ITGA5) were decreased in MMTV-Wnt1/Rarb-/-animals (Figure 3B). This observation was further confirmed by immunofluorescence showing higher E-cadherin levels in sections obtained from Rarb-/- mice (Figure 3C), as well as increased translocation of β-catenin into the nucleus in wild type cells (Figure 3D). Collectively, these data indicate that Rarb ablation results in suppression of EMT in the Wnt1-induced tumors.

Inhibition of epithelial-mesenchymal transition in Wnt1-induced Rarb-null tumors.
Down regulation of the IGF1/AKT axis in Wnt1-induced Rarb-null tumors
IGF-1 plays a central role in cell growth, differentiation, survival, transformation and metastasis, and deregulation of the IGF signaling pathway is well recognized as a key contributor to the progression of multiple cancers including mammary gland tumors. The IGF-1 signaling axis can also affect EMT. IGF-1 binds IGF1R to induce PI3K and activates AKT to repress GSK3β. GSK3β is a repressor of β-catenin and Snail able of inhibiting E-cadherin expression and subsequently promotes EMT and tumor progression. While we observed a reduction in the expression of Igf1 in the tumor compartment, IGF-1 is also produced by stromal cells. Western blotting using protein lysates obtained from whole tumors showed a decrease in endogenous IGF-1 levels and an increase in the expression of IGFBP5, an antagonist of IGF signaling, in the Rarb-null samples (Figure 4A). We next investigated whether the reduced levels of IGF-1 had an impact on AKT expression and activity in the tumors. Indeed, tumors obtained from MMTV-Wnt1/Rarb-/- animals displayed a small decrease in total AKT levels and a sharp reduction of phosphorylated AKT at serine 473 (Figure 4A). In addition, we found increased phosphorylation of GSK3β and decreased expression of Snail, both of which are downstream components of the IGF-1/AKT axis (Figure 4A). While no significant difference in the serum concentration of IGF-1 between Rarb-null animals and their wild type siblings was detected by ELISA (Figure 4B), we found significantly lower IGF-1 concentrations in the supernatants collected from Rarb-null CAFs in culture compared to the supernatants obtained from wild type fibroblasts (Figure 4C). These results indicate that changes in local IGF-1 levels are not due to a systemic effect of the absence of RARβ but intrinsic to the tumor microenvironment. Furthermore, retinoic acid induced the expression of IGF-1 mRNA from wild type fibroblasts (Figure 4D). Finally, tumor cells isolated from the Wnt1-induced tumors in wild type mice were cultured in the presence of conditioned media obtained from Rarb-null and wild type CAFs to measure growth rate. Wnt1-induced tumor cells cultured in the presence of conditioned media derived from Rarb-null fibroblasts showed a significant decreased cell growth rate (Figure 4E). In addition, immune-neutralization of IGF-1 in the conditioned medium derived from wild-type fibroblasts also reduced cellular growth rate, whereas supplementation of IGF-1 to the conditioned medium obtained from Rarb-null fibroblasts reversed the slow growth phenotype (Figure 4E). Taken together, these data indicate that in the absence of RARβ, reduced expression of IGF-1 and activity of its downstream signaling pathway contribute to attenuate EMT in Rarb-null tumors.
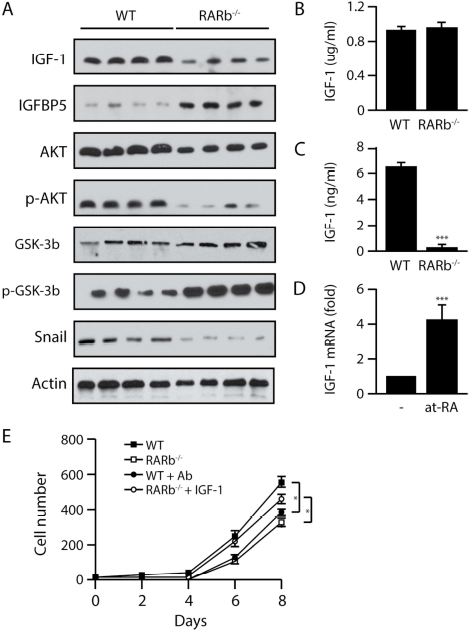
Down regulation of the IGF1/AKT axis in Wnt1-induced Rarb-null tumors.
Discussion
The antitumor activities of retinoids and their receptors have been investigated extensively and one of the receptors, RARβ, has been suggested to possess many of the functional characteristics of a tumor suppressor [13]. However, previous work from our laboratory showed that in the context of the whole organism, Rarb is not a tumor suppressor, but rather that its presence is required for the full oncogenic potential of Erbb2/neu [16]. Tissue recombination studies indicated that it is the absence of RARβ in the tumor microenvironment, rather than the status of Rarb in mammary epithelial cells, that affects the initiation and progression of Erbb2/neu-induced mammary gland carcinoma. In the current study, we monitored the influence of Rarb deletion on the induction of mammary tumors by the Wnt1 oncogene to test whether the absence of retinoid signaling via RARβ affects mammary tumor formation driven by a different oncogenic pathway. As previously observed with the MMTV-erbb2/neu model, ablation of Rarb in the MMTV-Wnt1 model showed extensive remodeling of the stroma during tumor progression through suppression of the activation and trans-differentiation of CAFs, reduction in matrix stiffness, decreased tumor angiogenesis and reduced inflammatory cell infiltration. Despite these similarities, the molecular mechanism underlining the interactions by which Rarb confers its effects on tumor cells is distinct between the two mouse models of human breast cancer. Clinically, these results imply that modulation of RARβ activity could have important applications to treat breast cancers of different cellular origin and/or driven by distinct oncogenic pathways.
In the Wnt1 model, oncogenic transformation has been proposed to target mammary gland stem cells or early progenitor cells. Therefore, Wnt1-induced tumors are morphologically composed of a variety of cell types, including distinct tumor luminal epithelial, basal/myoepithelial cells and host-derived stroma cells [26–28]. Wnt1 ligand produced by tumor cells can directly activate the surrounding stromal cells in a paracrine manner [29]. In return, stromal cells can affect tumor cells by expressing IGF-1, which activates IGF1R signaling in tumor cells [30]. This important molecular cross talk between tumor and stromal cells is severely weakened in the absence of RARβ. Deletion of Rarb down-regulated Wnt1 signaling as indicated by E-cadherin expression and nuclear localization of β-catenin resulting in lower IGF-1 expression in the stroma and suppression of the IGF-1/AKT axis in the tumor cells. Notably, reports demonstrated the importance of Wnt1 signaling in mammary stroma of human breast cancer which has been linked to EMT and poor clinical outcome [31–33].
Besides affecting stromal cells, Wnt1 ligand secreted by luminal epithelia can also target myoepithelial cells [29]. A recent report indicates that the Wnt1-driven tumor growth can be inhibited by a soluble Wnt receptor inhibitor [30]. The mechanism through which this effect is mediated involves the induction of IGFBP5, an antagonist of IGF signaling that has been shown to facilitate tumor growth by paracrine communication. The regulation of IGFBP5 is mediated by the β-catenin-dependent Wnt pathway. Strikingly, we observed that Igfbp5 was up-regulated about 2-fold in transcript levels while Igf1 was down regulated in the Rarb-null tumors. It has also been reported that overexpression of IGFBP5 results in an increase in apoptotic cells in the mammary glands, whereas mice with mutant Igfbp5 exhibit a decrease in apoptotic cells and a delay in mammary gland involution [34–36]. IGF-1 is a known growth and survival factor in mammary epithelial cells, and hyperactive IGF-1 signaling is found in various human tumors including breast cancer [37, 38]. Moreover, IGF1R activation has been shown to induce EMT by a PI3K/AKT-mediated mechanism and a concomitant increase in the expression of Snail and suppression of E-cadherin expression [39–44]. In agreement with these reports, we observed increased and decreased levels of E-cadherin and Snail proteins, respectively, as well as decrease cell proliferation and more apoptosis in Rarb-null tumors. Accordingly, both suppression of Wnt1 signaling pathway and repression of the IGF1/AKT axis in the absence of Rarb have an important negative impact on CAF activation and EMT in the Wnt1-induced tumors.
Conclusions
The failure of retinoid-based therapies to treat breast cancer led us and others to hypothesize that retinoic acid receptor isoform might have distinct and perhaps antagonistic functions in mammary gland development and in oncogenesis. Indeed, while activation or inhibition of the RARα isoform in mice are consistent with the anti-proliferative action of this receptor [17, 45], the activity of the RARβ and RARγ suggests that these isoforms act as proto-oncogenes in the context of the whole gland [16, 45]. In this work, we extended these findings by demonstrating that Rarb is crucial for the interaction between tumor cells and stromal cells. We found that Rarb expression has an important impact on Wnt signaling in both tumor and stromal cells. Specifically, inactivation of Rarb resulted in suppression of Wnt1 signaling which further inhibited the IGF-1/AKT/Snail/E-cadherin pathway and consequently repressed EMT (Figure 5). Since Wnt1-induced tumors in the mouse resemble triple negative breast cancer or basal-like breast cancer in which the Wnt and IGF-1 signaling pathways are known to be activated, this work suggests that specific RARβ antagonists could have useful clinical application in the treatment of poor outcome breast cancer.

Rarb is crucial for the interaction between tumor cells and stromal cells.
Public Datasets
Gene Expression Omnibus
GSE56391
Footnotes
Acknowledgements
This work was supported in part by grant DK043093 to MRS from the National Institutes of Health. Gene expression microarray data were generated by the Genomics Core Facility and analyzed with assistance from the Biostatistics Core Facility of the USC Norris Comprehensive Cancer Center, supported by Cancer Center Support Grant P30CA014089 from the National Cancer Institute. RC and D-YW were supported in part by National Institutes of Health-funded Training Grants T32 CA009320 and T32 GM067587, respectively.