
Abstract
The inhibitor of apoptosis protein survivin has long been of interest in the cancer literature for its role in both the regulation of cell proliferation and the inhibition of apoptosis. A growing body of literature has implicated survivin in the maladaptive pathways following vascular injury and, in particular, in the growth of vascular smooth muscle cells that comprise the hyperplastic neointimal lesions that characterize midterm vein bypass graft failure and restenosis following angioplasty and stenting. This review focuses on the emerging role of survivin in the regulation of smooth muscle cell growth and its implications for the prevention of restenosis following revascularization procedures. The expression, regulation, and function of survivin are addressed, as well as the current state of understanding regarding the effects of survivin inhibition in vitro and in vivo.
As a primary cause of in-stent stenosis and midterm bypass graft stenosis, the intimal hyperplasia lesion is composed primarily of smooth muscle cells bearing a proliferative, synthetic, and “dedifferentiated” phenotype. It is believed that this phenotypic switch from a differentiated, contractile phenotype occurs in the setting of increased cellular stress mediated by elevated local levels of inflammatory cytokines and growth factors as part of the vascular injury response. These conditions of cellular stress are thought to disrupt the balance between mitosis and apoptosis (programmed cell death) in vascular smooth muscle cells (VSMCs). Understanding the shift toward VSMC growth and survival is of fundamental importance to developing therapeutic interventions for the prevention and treatment of intimal hyperplasia. Survivin is a cell regulatory protein that has been extensively studied in cancer, particularly because it has significant roles in both permitting proliferation and preventing apoptosis. Recent studies have shown that survivin is intimately involved in the response to vascular injury and in intimal hyperplasia largely through its effects on smooth muscle cell growth.
Survivin Expression, Regulation, and Function
Survivin is a member of the inhibitor of apoptosis (IAP) gene family. It is a 1,619 bp gene that encodes a 16.3 kDa protein. 1 Virtually undetectable in terminally differentiated adult tissue, it is present during fetal development and is markedly upregulated in transformed cell lines and most human malignancies. 2 Survivin contains a single baculovirus IAP repeat, the functional subunit of the IAP family, which acts as an evolutionarily conserved inhibitor of caspases, the effector enzymes of apoptosis. 3 In the setting of cell stress, mitochondrial stores of survivin are released, resulting in blockade of the intrinsic pathway of apoptosis through inhibition of caspase 9. 4
Survivin is unique among other IAPs in that it exhibits a highly regulated pattern of expression, which involves both cell cycle–dependent and cell cycle–independent mechanisms. Cell cycle–dependent expression is involved in the regulation of mitosis and is controlled at the level of transcription. 5,6 Following its dramatic upregulation at G2/M, survivin localizes to centrosomes and to microtubules of the metaphase and anaphase spindle, playing a critical role in the faithful execution of mitosis. 7,8 Survivin is subsequently rapidly degraded by ubiquitination followed by proteosome-mediated destruction, which keeps levels low following mitosis. 9 Survivin also interacts with the tumor suppressor gene product p53 and its downstream target P21 through a balanced counterregulatory pathway that contributes to the cell cycle checkpoint function of both genes. 10,11 Inhibition of cell cycle–dependent survivin expression results in multiple mitotic defects and generation of multinucleated, polyploid cells. 12–14 Consistent with this prominent role in mitosis, survivin knockout mice demonstrate 100% lethality by embryonic days 3.5 to 4.5, with absence of mitotic spindles and failure of cell division. 15
The cell cycle–independent expression of survivin is predominantly associated with periods of cellular stress (Figure 1). This relationship has been extensively studied in the setting of malignancy where cells have significant and marked survival signaling in the setting of unfavorable (stressful) environments. Increased survivin expression in malignancy is considered a marker of more aggressive tumors and worse prognosis. 16–18 The cell cycle–independent expression of survivin acts by two mechanisms to promote cellular growth. First, when expressed in a cell cycle–independent fashion, the cell cycle regulatory function of periodic survivin expression, as described above, is lost, leading to unchecked proliferation. Second, with the persistent upregulation of survivin, the cell becomes markedly resistant to apoptosis. Indeed, in experimental models, survivin expression modulates cell proliferation and survival in vitro and in vivo. Upregulation of survivin inhibits cell death in multiple cell lines, 19,20 and transgenic expression of survivin in keratinocytes in vivo demonstrates resistance to apoptosis after ultraviolet B irradiation. 21 In contrast, molecular antagonists of survivin (antisense, ribozymes, ribonucleic acid [RNA] interference, or dominant-negative mutants) cause caspase-dependent cell death and sensitize cells to apoptotic stimuli. 19,22 Recent work has demonstrated a relationship between cell cycle–independent survivin expression and its molecular chaperone heat shock protein 90 (HSP90). Targeted disruption of the survivin–HSP90 interaction leads to rapid cell death via apoptosis in multiple cancer cell lines. 23,24 The dependence of cell cycle–independent survivin function on a stress protein (HSP90) further emphasizes the role of survivin in mediating cell survival under stress conditions.
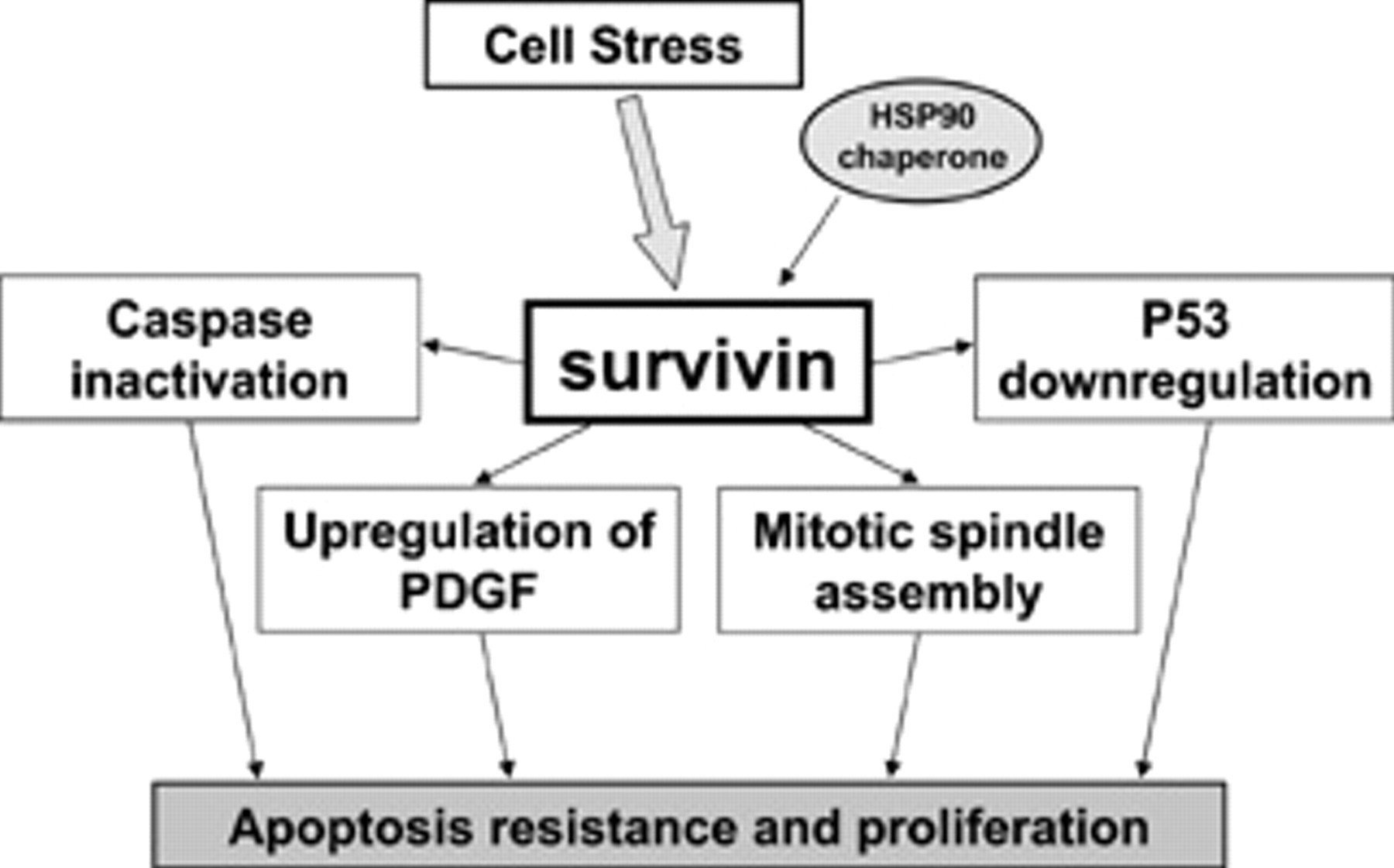
General schema of survivin function. Survivin interacts with multiple regulatory processes of cell survival and proliferation. In the setting of cellular stress, upregulation of survivin allows unchecked proliferation with concomitant resistance to apoptotic stimuli. PDGF = platelet-derived growth factor.
Survivin Expression in Vascular Injury
Survivin expression (at the protein and RNA level) is virtually undetectable in normal arteries and veins in mice, rabbits, and humans. In contrast, striking upregulation of survivin occurs in vessels subjected to various injury mechanisms, including wire injury to the mouse femoral artery, balloon arterial injury or vein grafting in the rabbit (Figure 2A), diet-induced atherosclerotic lesions in the rabbit, and human vascular lesions (including atherosclerotic carotid endarterectomy plaques and stenotic lower extremity vein bypass grafts). 25,26 Following vascular injury, survivin expression in VSMCs is associated with α-actin-positive and smoothelin-negative staining consistent with the dedifferentiated phenotype of “myofibroblast” cells. 26 RNA analysis of arteries and vein grafts from rabbits confirmed that survivin upregulation occurred at the level of transcription (Figure 2B), beginning several days to a week after the vascular injury and remaining persistently elevated up to 3 months later. 26 These initial, in vivo observations established that survivin is a molecule of clear relevance to vascular lesion formation.
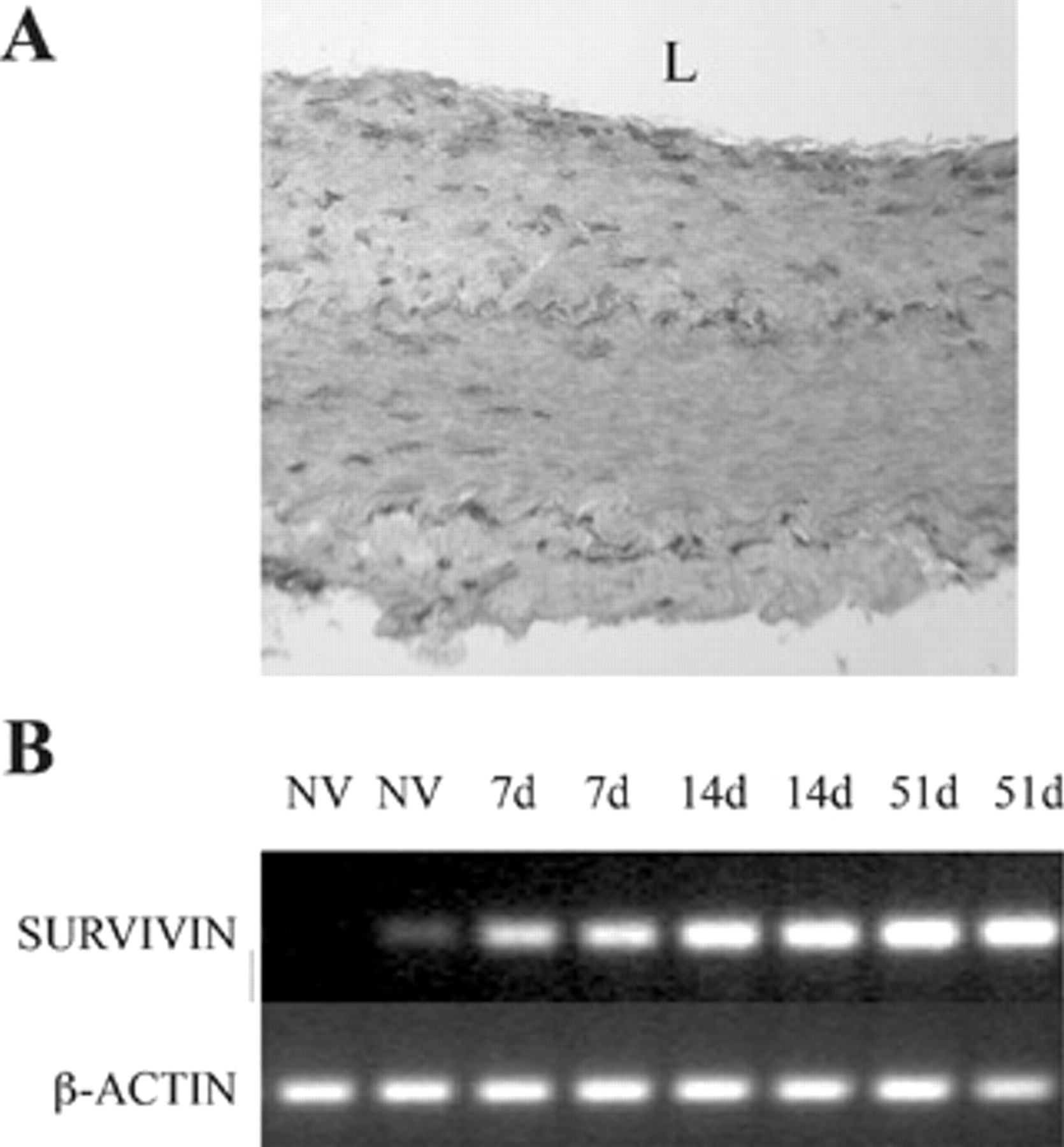
Survivin is upregulated after vascular injury in vivo. A, Representative immunostaining for survivin in a rabbit model of vein bypass reveals transmural upregulation of intracellular survivin at post-operative day 14 (100× magnification). Anti-survivin primary antibody was detected using avidin-biotin-peroxidase (NovaRed, Vector Laboratories, Burlingame, CA). B, Representative semi-quantitative reverse-transcriptase polymerase chain reaction of tissue lysates from rabbit bypass grafts showing early and persistent upregulation of survivin at the level of transcription. (“NV” denotes normal vein and “7d”, “14d”, and “51d” refer to the postoperative day at the time of bypass graft harvest.)
Survivin Regulates VSMC Growth and Gene Expression
The mediators of survivin upregulation and the mechanisms by which survivin may influence VSMC phenotype are being intensely investigated. Initial studies have demonstrated that, in vitro, increased survivin expression is associated with an array of growth factors and cytokines that are independently upregulated after vascular injury, including angiotensin II, tumor necrosis factor α, 27 heparin-binding epidermal growth factor, and platelet-derived growth factor (PDGF). 25,28
The effect of survivin expression and inhibition has been subsequently studied using an adenovirus transduction vector to deliver either wild-type survivin (Ad-SVV) or a nonfunctional mutant encoding a Thr34→Ala survivin mutant (Ad-T34A), which abolishes an essential phosphorylation site for p34cdc2 kinase. The Ad-T34A construct thus codes an unstable dominant negative mutant that competitively inhibits endogenous survivin effects. 29 Transduction of primary VSMC with Ad-SVV directly induced a proliferative response, which was serum dependent (3.2-fold increase in 10% serum vs 1.5-fold in 0.5% serum). In contrast, infection with Ad-T34A markedly inhibited proliferation in all serum concentrations. 28 This confirms an essential role for intact survivin function in VSMC growth in vitro (Figure 3).
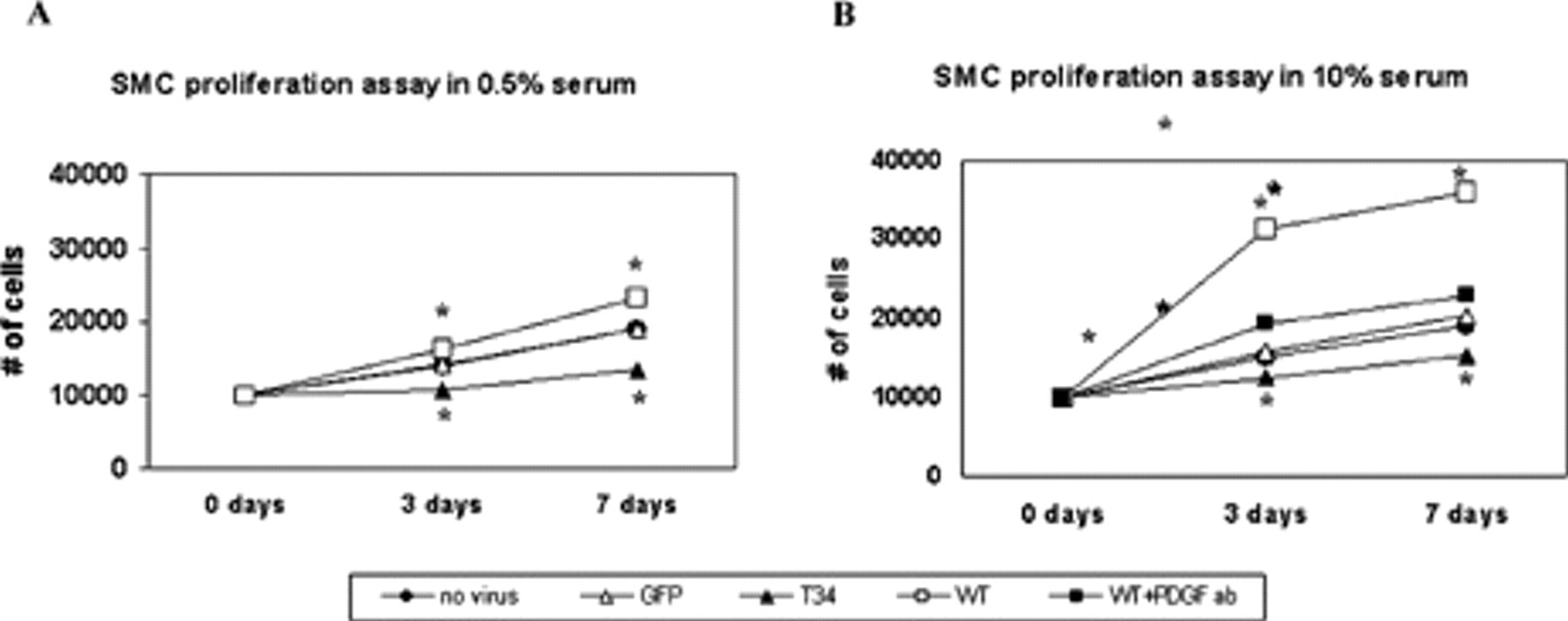
Vascular smooth muscle cell (VSMC) proliferation and apoptosis resistance are altered by survivin expression. Overexpression of survivin (denoted as “WT”) via adenoviral transduction (Ad-SVV) increases proliferation of VSMCs in comparison with untreated cells (denoted as “no virus”) and those transduced with an irrelevant control gene (Ad-GFP, denoted as “GFP”). This effect is abrogated both by transduction with a dominant negative survivin construct (Ad-T34A, denoted as “T34”) and by treatment with an anti–platelet-derived growth factor (PDGF) neutralizing antibody (denoted as “WT + PDGF ab”). See text for quantification; *p < .05 for Ad-SVV and Ad-T34A versus Ad-GFP. Adapted with permission from online supplement to Wang et al. 28
Survivin expression also markedly increases the apoptotic threshold of VSMCs. Transduction of VSMCs with Ad-SVV protects them from apoptosis by either ceramide or cytokines. 25 Conversely, transduction with Ad-T34A directly induces apoptosis in the absence of cytokine and greatly sensitizes cells to cytokine-mediated apoptosis (Figure 4). 28
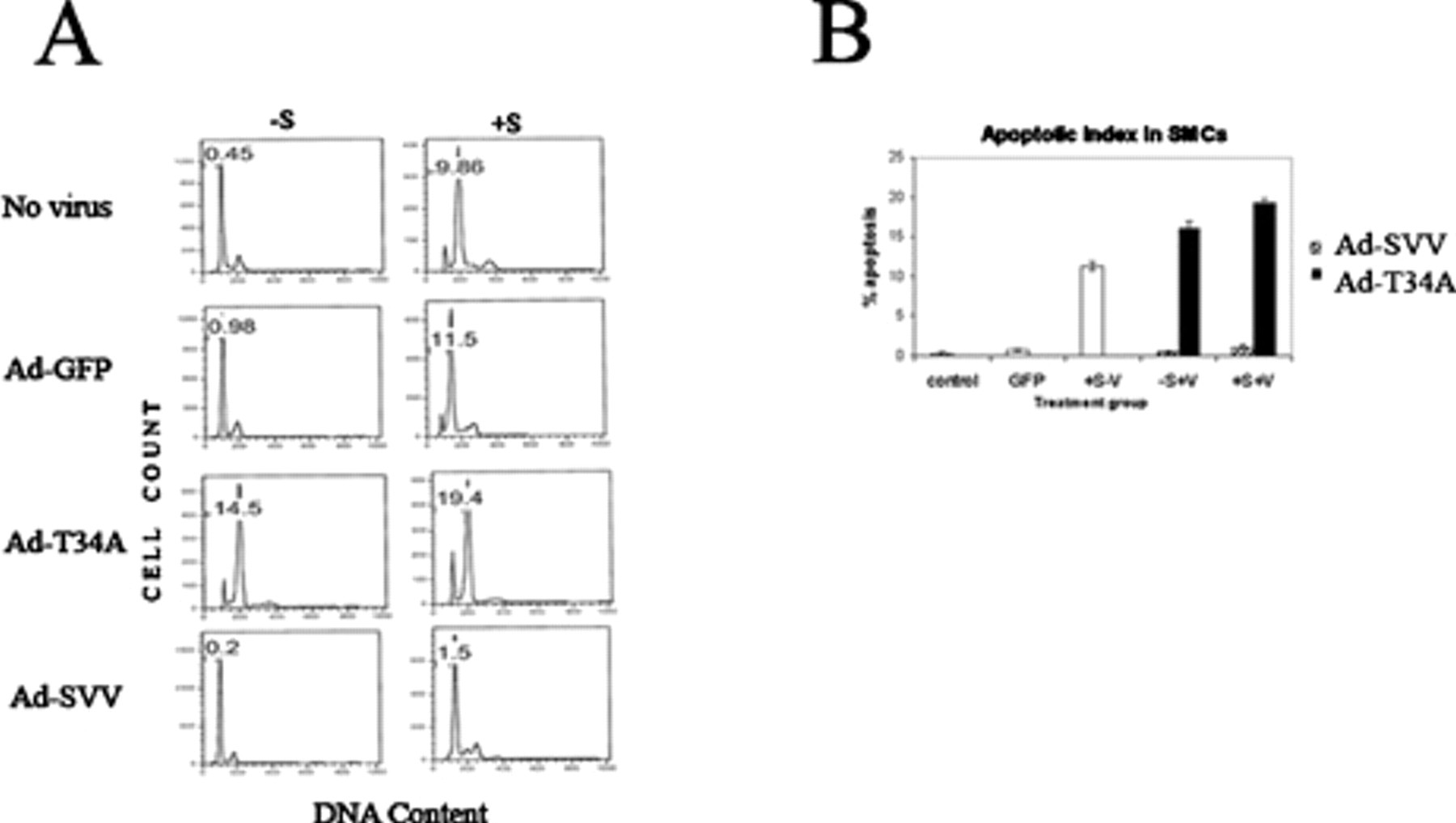
Representative flow cytometry histograms (A) and summary plot (B) of deoxyribonucleic acid (DNA) content analysis of vascular smooth muscle cells (SMCs), treated with Ad-GFP (transduction control), Ad-SVV, or Ad-T34A (+V), in the presence or absence of apoptosis-inducing cytokine stimulation (± S). Number indicates percentage of apoptotic cells. Adapted with permission from online supplement to Wang et al. 28
As an extension of this work, the role of PDGF, an established mediator of intimal hyperplasia, in relation to survivin has been further examined. As noted above, PDGF induces survivin expression in VSMCs. In addition, the known cytoprotective effects of exogenous PDGF were eliminated in cells transduced with Ad-T34A. Furthermore, blockade of PDGF using a neutralizing antibody that recognizes all PDGF isoforms markedly reduced (85%) the proliferative effects of survivin overexpression. These results establish a dynamic relationship between exogenous PDGF and cell cycle–independent survivin function. However, survivin has also been shown to induce PDGF-A and PDGF-B promoter activity by transduction with Ad-SVV. This induction is suppressed by transduction with Ad-T34A. 28 Taken together, these results demonstrate not only that PDGF-induced survivin expression promotes dysregulated VSMC growth and survival but that this pathway may be embedded in a positive feedback loop involving PDGF.
Survivin Regulates Vascular Repair and Remodeling In Vivo
The in vitro effects of survivin on VSMCs have been translated into models of vascular injury in which disruption of survivin signaling significantly reduced neointima formation. In arterial injury models, exposure of the injured vessels to Ad-T34A resulted in significant reductions in neointima formation. 25,26 Extending this methodology to a model of vein bypass in rabbits, vein grafts treated with a single adventitial application of Ad-T34A demonstrated a significant reduction (41%) in wall thickness. In contrast, vein grafts treated in an identical manner with Ad-SVV demonstrated a marked increase (137%) in wall thickness. 28 Therefore, the vascular injury response and subsequent neointima formation in arterialized veins are inhibited by disruption of survivin signaling, whereas overexpression of survivin leads to a marked increase in graft thickness.
The effect of a single application of Ad-SVV and Ad-T34A at the time of bypass also suggests that survivin-mediated remodeling of the vessel wall is established early in the time course of the vascular injury response. 28 Indeed, evaluation of early time points following bypass revealed a 2.75-fold increase in apoptotic index (by TUNEL assay) in grafts treated with Ad-T34A in comparison with a 0.66-fold increase (net decrease) in grafts treated with Ad-SVV. At the same time point, the proliferative index was reduced (by Ki-67 staining) in grafts treated with Ad-T34A (60% of control), whereas proliferation was markedly increased in grafts treated with Ad-SVV (275% over control). Similar findings have also been demonstrated in a rat model of pulmonary hypertension, in which treatment with Ad-T34A induced VSMC apoptosis, reduced proliferation, and reversed hypertrophic changes in the pulmonary artery in vivo. 30 These findings support the hypothesis that early alteration of the VSMC survival and proliferation program following vascular injury has significant implications for the degree of intimal hyperplasia at late time points.
Therapeutic Interventions for Modulating Survivin Effects in VSMC Growth
Taken together, the clear effects of survivin on VSMC growth in vitro and the corresponding effects in animal models of vascular injury and vascular bypass warrant exploration of therapeutic interventions that modulate the survivin-mediated response to vascular injury and remodeling. In addition to RNA interference and virus-mediated delivery of dominant negative mutants, additional methods of survivin inhibition are also being explored. Our research group has shown that 3-hydroxy-3-methylglutaryl coenyzme A reductase inhibitors (“statins”), which are proapoptotic for VSMCs in vitro, 31,32 act through a survivin-mediated pathway. Human VSMCs that were exposed to lovastatin (100 μM) underwent apoptosis, an effect that was eliminated with the addition of mevalonate. However, with survivin overexpression via Ad-SVV, the apoptotic effect of lovastatin was eliminated (Figure 5). In a subsequent organ culture model using segments of rabbit aorta maintained for up to 14 days in media containing 30% serum, lovastatin treatment (1–50 μM) produced a dose-dependent attenuation of wall thickness (up to 40%) that was highly associated with a reduction in survivin expression (not shown). Taken together, these data suggest that the proapoptotic effects of statins on VSMCs depend on an inhibition of survivin and, conversely, that upregulated survivin expression renders VSMCs resistant to these effects. These results are supported by a recent report that found a correlation between statin-induced apoptosis and decreased levels of survivin in VSMCs. 33

Lovastatin-induced apoptosis in vascular smooth muscle cells (SMCs) is eliminated by overexpression of survivin. Summary plot of cells transduced with Ad-GFP (transduction control) or Ad-SVV and treated with lovastatin or lovastatin + mevalonate. Apoptotic index measured by fluorescence-activated cell sorter for DNA content.
Another avenue of current investigation involves disruption of the HSP90–survivin interaction in VSMCs. These experiments, analogous to published results in the cancer literature, demonstrate an increase in apoptosis of VSMCs with inhibition of HSP90 chaperone function. This effect is preceded by loss of functional HSP90 client proteins, including survivin.
Conclusions and Future Directions
VSMCs have marked alterations in proliferation and survival following vascular injury. These alterations lead to intimal hyperplasia and restenosis from proliferation of dedifferentiated VSMCs on the luminal side of the vessel wall. As a key regulator of both proliferation and cellular survival, survivin has been demonstrated to have a key role in VSMC growth in the setting of vascular injury and remodeling. The cell cycle–independent expression of survivin markedly modulates VSMC survival and proliferation, leading to significant alterations in cellular phenotype. The relevance of survivin to vascular injury and VSMC phenotype has led to ongoing exploration of therapeutic interventions that can modulate these signals.