
Abstract
Chemokines are critical for white blood cell recruitment to injured tissues and play an important role in normal wound healing processes. In contrast, impaired wound healing in diabetic patients is accompanied by decreased early inflammatory cell infiltration but persistence of neutrophils and macrophages in the chronic, nonhealing wounds. These changes in inflammatory cell recruitment occur in conjunction with alterations in chemokine and growth factor expression. In addition to leukocyte trafficking, many different cell types, including endothelial cells, fibroblasts, and keratinocytes, produce and respond to chemokines, and these interactions are altered in diabetic wounds. Thus, the chemokine system may have both direct and inflammatory-mediated effects on many different aspects of diabetic wound healing. The potential roles of chemokines and inflammatory or immune cells in nonhealing diabetic wounds, including impairments in growth factor expression, angiogenesis, extracellular matrix formation, and reepithelialization, are examined.
Nonhealing wounds constitute one of the most frequent medical problems of patients with diabetes. 1 The incidence of foot ulcers among diabetic patients is estimated to be 2% per year, with an average cost of $27,987 (1999 data) per patient for 2 years of treatment. 2 In addition, the chronicity of diabetic foot ulcers and associated complications are significant risk factors for limb amputation. 2–4 Previously, the role of inflammation and chemokines have been appreciated in the pathogenesis of diabetic and nonhealing wounds. 1,5 To expand therapeutic options that can potentially promote diabetic wound healing, a better understanding of the complex cellular interactions occurring in diabetic wounds is imperative. This article examines the role of chemokines in chronic diabetic wounds. The general functions of chemokine are reviewed, followed by a brief description of normal wound healing with a more in-depth analysis of the potential roles of chemokines and inflammatory or immune cells in nonhealing diabetic wounds, including impaired growth factor expression, angiogenesis, extracellular matrix formation, and reepithelialization defects.
Brief Review of Chemokines
Chemokines (or chemotactic cytokines) are small heparin-binding proteins that direct the movement of circulating leukocytes to sites of inflammation or injury via interaction with specific membrane-bound receptors and, as such, contribute to the pathogenesis of a variety of diseases. 6 To date, approximately 50 human chemokines have been identified and are segregated into four families based on chemical structure and function. Depending on the spacing or presence of four conserved cysteine residues, chemokines are classified into CC, CXC, CX3C, and XC families. 7 Chemokines act through receptors belonging to the superfamily of rhodopsin-like G protein–coupled 7-transmembrane receptors. 8 Although receptor–ligand interactions are predominantly family specific, there is considerable functional redundancy and promiscuity among chemokines and their receptors within families. 8
CXC chemokines primarily attract neutrophils and lymphocytes 9 and are believed to orchestrate the early phases of wound healing. 6 CXC chemokines are further subdivided based on the presence or absence of a glutamic acid-leucine-arginine (ELR) motif immediately before the first cysteine residue. 10 Whereas ELR+ CXC chemokines promote angiogenesis, ELR− chemokines are angiostatic. 8 With normal wound healing, neutrophil infiltration induced by CXC chemokines is followed by mononuclear cell recruitment in response to CC chemokines. 6 Thus, a well-ordered pattern of inflammatory cell infiltration promotes effective wound healing and occurs in response to a coordinated expression of chemokines. 11–13 Whereas CXC and CC chemokines have received considerable attention, CX3C and XC chemokines remain poorly defined but play significant roles in immune-mediated conditions such as atherosclerosis, rheumatoid arthritis, immunoglobulin A nephropathy, and tumor formation. 6
Normal Wound Healing and Alterations of Inflammation in Diabetic Wounds
The normal healing process can be defined by a number of overlapping events: clot formation, inflammation, reepithelialization, angiogenesis, granulation tissue formation, wound contracture, scar formation, and tissue remodeling. 5,14 Diabetic wounds are characterized by functional defects in the majority of these events, leading to impaired wound healing, in addition to local ischemia caused by well-recognized macro- and microvascular occlusive disease. 5 Thus, the etiology of nonhealing diabetic wounds is multifactorial, with significant derangements in the coordinated reparative process involving resident and infiltrating cell populations.
Following local tissue trauma, inflammatory or immune cells consisting of neutrophils, macrophages, mast cells, and lymphocytes sequentially infiltrate the wound site and not only serve as immunologic effectors but also produce potent growth factors and cytokines. 15 Impairments in diabetic wound healing exhibit decreased growth factor expression in tissues (reviewed by Medina and colleagues 1 ). Central to the coordinated recruitment of leukocytes into injured tissues are chemokines. 6 Chemokine receptors have been identified on multiple complementary cell populations such as endothelial cells, 8 vascular smooth muscle cells, 16 fibroblasts, 15 and keratinocytes, 17,18 suggesting that chemokines may contribute directly toward angiogenesis, extracellular matrix remodeling or formation, and reepithelialization.
As described above, the appropriate influx and activation of inflammatory cells within sites of injury promote effective wound healing by clearance of necrotic debris, neutralization of noxious stimuli, and growth factor production, 19 as well as stimulation of angiogenesis. 20 Wounds of diabetic patients demonstrate impairments not only in the initial recruitment of inflammatory cells following injury 21 but also in the bactericidal activity of neutrophils and other leukocytes. 22,23 Once infiltration is finally established, wounds of genetically diabetic (db/db) mice retain a high concentration of neutrophils and macrophages, leading to sustained increases in the proinflammatory cytokines interleukin (IL)-1β and tumor necrosis factor α. 13 In turn, persistent neutrophil and macrophage infiltration is associated with sustained expression of chemokines such as macrophage inflammatory protein 2 (MIP-2/CXCL8) and monocyte chemotactic protein 1 (MCP-1/CCL2), potent chemotactic agents for neutrophils and macrophages, respectively. 13 Interestingly, sustained expression of MIP-2 and MCP-1 is associated with differential localization of neutrophils and macrophages at the wound bed compared with that in control mice. 13 Thus, not only are the quantity and functional properties of individual inflammatory cell populations important for effective wound healing, but the temporal relationship between infiltrating cells appears to be critical as well. 13 In conclusion, diabetic wounds demonstrate initial impairments in leukocyte recruitment following injury; however, once established, persistent leukocyte infiltration causes an inappropriately prolonged inflammatory response, potentially impeding normal wound healing.
Diabetic Wounds and Growth Factor Expression
Growth factors influence cellular proliferation and function, induce migration of inflammatory cells into the wound bed, and stimulate protein synthesis or inhibit these events as healing progresses. 5 The tissue level and expression kinetics of growth factors may dictate their in vivo effect. 24 Indeed, nonhealing diabetic wounds are characterized by alterations in the finely balanced expression of various growth factors, including transforming growth factor β, 25 insulin-like growth factor 1, 26 platelet-derived growth factor, 27 nerve growth factor, 28 keratinocyte growth factor, 29 IL-6, 30 and vascular endothelial growth factor (VEGF). 31 The exact etiology of relative growth factor deficiencies in diabetic wounds is difficult to discern given the presence of multiple cellular sources of growth factors within proliferating wounds, including the persistence of inflammatory cells. Although growth factors from cell types such as keratinocytes, fibroblasts, and endothelial cells can direct continued proliferation and protein synthesis and may be directly affected by chemokines, macrophages, through growth factor secretion, are believed to serve as the initiators of the proliferative process in normal circumstances. 24 The suggested role of chemokines in promoting macrophage-dependent growth factor production in human wounds is indirect and limited to inflammatory cell recruitment and activation as direct stimulation of isolated macrophages in vitro with various concentrations of MCP-1 was unable to stimulate growth factor production. 12 Thus, time-sensitive and coordinated recruitment of macrophages into the site of injury may be necessary for effective wound healing.
Diabetic Wounds and Angiogenesis
In normal wounds, endothelial cells promote healing by mediating and regulating the recruitment of inflammatory cells into sites of injury and the establishment of a capillary network, or angiogenesis. Chemokines are important in inflammatory cell recruitment and angiogenesis. 15 Expression of chemokines such as IL-8 and MCP-1 on endothelial cell surfaces facilitates transmigration of leukocytes from the intravascular compartment into sites of inflammation. 32,33 As such, nonhealing diabetic wounds are characterized by decreased leukocyte infiltration acutely following injury. 21 Interestingly, diabetic wounds exhibit increased and sustained induction of MIP-2 and MCP-1 levels within injured tissue, 13 suggesting alternative mechanisms for decreased inflammatory infiltration. Nonetheless, once within injured tissues, inflammatory cells promote reestablishment of a capillary network by production of angiogenic factors such as VEGF, 20 and nonhealing diabetic wounds express decreased tissue levels of VEGF. 31 Interestingly, induction of VEGF expression restores angiogenesis in diabetic mice and accelerates wound healing. 34 Furthermore, inflammatory cells promote angiogenesis through clearance of necrotic tissues and extracellular matrix digestion, 35 as well as possible incorporation into the vessel wall. 36
The role of chemokines in angiogenesis is exerted both indirectly, through inflammatory cell recruitment, but also directly, through interactions with chemokine receptors on the endothelium. Although controversial, endothelial cells express receptors for CXC and CC chemokines promoting angiogenesis through endothelial cell proliferation and migration. 37,38 Specifically, the direct angiogenic properties of MCP-1 are attributed to upregulation of hypoxia-inducible factor α, which, in turn, induces VEGF-A expression. 39 In addition, bone marrow–derived endothelial progenitor cells express CXCR4, promoting angiogenesis through incorporation into capillary networks and/or by paracrine effects from endothelial progenitor cell–secreted factors. 40–42 Thus, the etiology of abnormal angiogenesis in diabetic wounds occurs through multiple mechanisms. 43
Diabetic Wounds and Extracellular Matrix
Extracellular matrix functions in wound repair as a scaffold for proper cellular attachment and function. Moreover, extracellular matrix integrity is maintained by the closely balanced production and degradation of extracellular proteins such as collagen and glycosaminoglycans. Diabetic wounds display both impairments in extracellular matrix production 44 and increased degradation owing to elevated concentrations of proteolytic matrix metalloproteinases (MMPs). 45 The overall effect of decreased collagen and glycosaminoglycan content within diabetic wounds is associated with decreased wound strength and an impaired ability to withstand minor trauma. 5
Chemokines are influential in both production and degradation of extracellular matrix. Chemokine-dependent inflammatory cell recruitment and subsequent growth factor secretion promote extracellular matrix production. 46 However, fibroblasts both produce and respond to chemokines. 15 Following in vitro chemokine-induced stimulation, fibroblast collagen production 47 and migration 48 are altered. Moreover, gene expression of MMP-1 and the tissue inhibitor of metalloproteinase 1 (TIMP-1) is enhanced in isolated human dermal fibroblasts following MCP-1 stimulation. 49 Although chemokines can induce both profibrotic and proteolytic functions in vitro, the normal MMP and TIMP balance is shifted toward extracellular matrix degradation in chronic, nonhealing diabetic wounds, 45 underscoring the delicate balance needed for effective wound repair.
MMPs are produced by different cell types, such as keratinocytes, fibroblasts, endothelial cells, neutrophils, and macrophages. 45 Fluid from chronic wounds impairs cell proliferation and angiogenesis and exhibits increased levels of MMPs. 50 The etiology of this phenomenon may be explained by direct chemokine-induced MMP production by resident cells within the wound (ie, keratinocytes, fibroblasts), as well as by the sustained inflammatory cell infiltrate. Thus, chemokines can exert both direct and inflammatory-mediated effects on the extracellular matrix.
Diabetic Wounds and Reepithelialization
Reepithelialization of a wound begins within hours after injury by rapid reestablishment of an epithelial barrier by keratinocyte migration over a provisional matrix. 51–53 In contrast, nonhealing diabetic wounds demonstrate impairments in reepithelialization and provisional matrix deposition critical for effective wound healing. 54–57 In particular, chronic wounds contain multiple proteases that digest fibronectin and growth factors within the fibrin clot, resulting in a corrupt provisional matrix that no longer supports reepithelialization or granulation tissue formation. 55,56 Reepithelialization of wounds may be directly affected by both CXC and CC chemokines as well. Human keratinocyte migration 18 and proliferation 58 in vitro are stimulated by IL-8 via CXCR2. In addition, in vivo studies confirm significant delays in reepithelialization in CXCR2−/− mice. 59 MCP-1 is produced by keratinocytes and is upregulated during normal wound healing. 12 Moreover, significant delays in wound reepithelialization are observed in mice deficient in MCP-1. 60 Thus, impairments in diabetic wound reepithelialization may be promoted by provisional matrix derangements and chemokine-dependent keratinocyte dysfunction.
Conclusions
Wound healing involves many complex, interrelated processes that involve multiple cell types. Chemokines, through both direct and inflammatory-mediated mechanisms, play an important role in the normal healing process, whereas dysregulation of chemokine expression may contribute to the pathogenesis of nonhealing wounds present in diabetic patients (Figure 1). A better understanding of the molecular mechanisms and cellular interactions, including the roles that chemokines play in these events, is critical for the development of novel therapeutic strategies to promote diabetic wound healing.
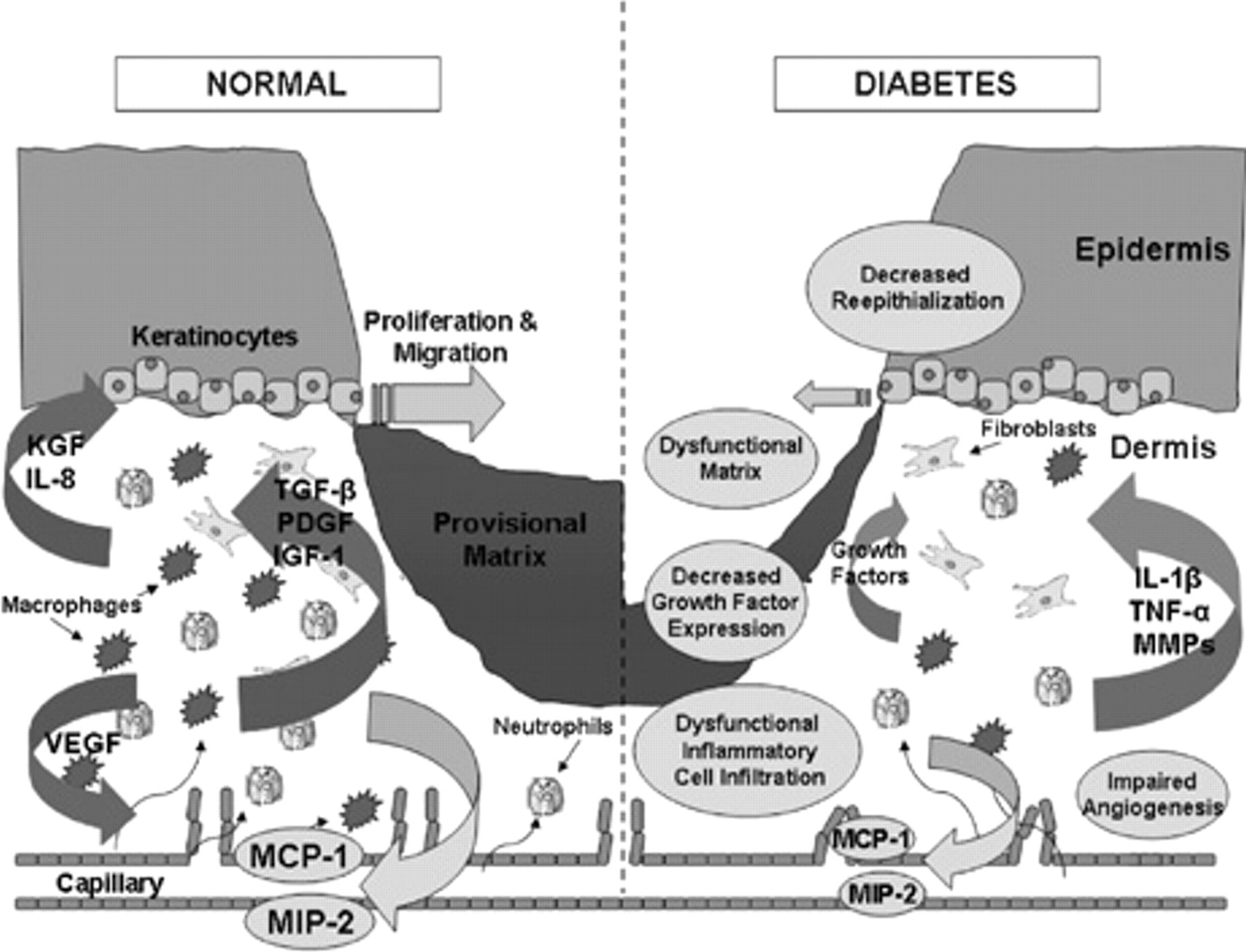
Normal wound healing and alterations in diabetic wounds. Schematic comparing various components of wound healing in normal versus diabetic patients. Under normal conditions, wound healing is characterized by effective inflammatory cell recruitment in response to chemokines macrophage inflammatory protein 2 (MIP-2) and monocyte chemotactic protein 1 (MCP-1), followed by production of multiple growth factors promoting matrix formation, angiogenesis, and reepithelialization. In contrast, diabetic wounds exhibit delayed inflammatory cell infiltration owing to decreased chemokine expression with decreased growth factor production, leading to impairments in angiogenesis, matrix formation, and reepithelialization. During later stages of diabetic wound healing, persistence of inflammatory cells within injured tissues causes continued damage and turnover through increased expression of interleukin-1β (IL-1β), tumor necrosis factor α (TNF-α), and matrix metalloproteinases (MMPs). IGF-1 = insulin-like growth factor 1; IL-8 = interleukin-8; KGF = keratinocyte growth factor; PDGF = platelet-derived growth factor; TGF-β = transforming growth factor β; VEGF = vascular endothelial growth factor.
Footnotes
Supported by grants from the National Institutes of Health (HL070158, HL074236) and the Veterans Administration.