
Abstract
Since its initial characterization in 1988, over 18,236 papers, including 2,485 reviews, have been published in the endothelin (ET) field. Over this period, several generations of selective and mixed (dual) ET receptor antagonists (ERAs), from peptidic backbones to orally active potent (subnanomolar) small molecular compounds, have been developed. These agents have been studied in many experimental animal models of various pathological conditions (cardiovascular, respiratory, and neuro-immunological). Continued basic research has led to a better understanding of the complex interactions between the ET axis and other biologic systems in human pathophysiology. The first clinical trial involved patients with idiopathic pulmonary arterial hypertension and led to approval of bosentan (Tracleer) for use in the United States and Europe in 2002. Since then, bosentan, the only currently approved dual (mixed) ERA, has been used in numerous other clinical trials. In addition, more selective ETA receptor antagonists (ambrisentan, atrasentan, avosentan, clazosentan, darusentan, and sitaxsentan) are undergoing clinical trials. Here we outline the ERAs undergoing development and summarize the standing of completed and ongoing trials at the time of the Ninth International Conference on Endothelin and even thereafter. This review is intended to provide a useful reference for those interested in the current state of clinical trials involving ERAs, and to identify lessons that might apply to the design of future trials.
Introduction
The first widely used endothelin (ET) receptor antagonists (ERAs) were BQ-123, a selective ETA receptor antagonist (ERA-A; Ref. 1), and BQ-788, a selective ETB receptor antagonist (ERA-B; Ref. 2), both from Banyu (Merck). We (3–6) and others (7–18) have reviewed the chemistry and pharmacology of these ERAs as well as the characteristics of other classes of blockers of the ET system, such as single ET-converting enzyme inhibitors or dual and triple vasopeptidase inhibitors incorporating ET-converting enzyme inhibitors (19, 20).
Currently, both BQ-123 and BQ-788 remain useful tools for defining the pathophysiology of the ET system; numerous studies have employed these agents. Unfortunately, their high cost and parenteral method of administration (peptidic nature) has precluded their use in large clinical trials (Table 1). Ro 47–0203 (bosentan) became the first ERA to undergo Phase I clinical trials, with subsequent studies demonstrating clear therapeutic benefit (efficacy) and safety, leading to Phase II–III clinical trials in patients with idiopathic pulmonary arterial hypertension (iPAH) and pulmonary arterial hypertension (PAH) secondary to connective tissue diseases. While the therapeutic applications for bosentan are expanding to address other related pathological conditions (such as secondary PAH), additional compounds have been chosen to also target PAH (ambrisentan and sitaxsentan). Further agents are aiming at resistant hypertension (darusentan and TBC3711), prostate cancer (atrasentan, ZD4054, and YM598), and cerebral vasospasm (clazosentan), as well as other disorders (bosentan, ambrisentan, and sitaxsentan).
Profile of Endothelin Receptor Antagonists Used in Preclinical Studies and Subsequent Clinical Academic Studies and Formal Trials
Table 1 summarizes the ERAs that have been used in several clinical academic studies and formal trials in healthy subjects and patients.
Peptidic ERA: BQ-123 and BQ-788.
These two ERAs (Table 1) are the most used and published to date. They
were developed by Banyu Pharmaceutical Co. (Merck) and are now commercially available.
Their chemistry, pharmacology, pharmacokinetics, and pharmacotoxicology are well described
elsewhere (42). BQ-788
([N-cis-2,6-dimethylpiperidinocarbonyl-
Dual (Mixed) ERA (In Alphabetical Order).
Bosentan.
The first of its class, the U.S. Food and Drug Administration–licensed Tracleer was developed at Hoffman-La Roche (43) and followed Martine and Jean-Paul Clozel when Actelion was founded. Bosentan (Ro 47–0203; (4-tert-butyyl-N-[6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2,2′-bipyrimidin-4yl] benzene sulfonamide)) is an orally available (50%), nonpeptidic, competitive antagonist of both the ETA and ETB receptor subtypes, thus designed as mixed (dual) ERA, with a half-life of 5.4 hrs. Of all the ERAs, bosentan has been tested in the greatest number of different preclinical experimental animal models of diseases (e.g., hypertension, heart failure, pulmonary hypertension, renal dysfunction, remodeling, end-organ damage, and cerebral vasospasm following subarachnoid hemorrhage) as presented in over 800 publications (Table 1). Furthermore, such preclinical studies have permitted head-to-head comparisons with other treatments, indicating the benefit of ERAs over conventional approaches. Other studies were conducted using human blood vessels. These studies have led to numerous clinical trials, which are summarized in Tables 2 and 4 (completed trials) and 6 (ongoing trials) and are discussed below. A number of reviews presenting the profile, progress, and current status of Tracleer have been published in recent years (63–69).
Enrasentan.
SB 217242 ((1S, 2R, 3S)-3-[2-(2-hy-droxyeth-1-yloxy)-4-methoxyphenyl]-1-(3,4-methylene-dioxphenyl)-5-propoxyindane-2-carboxylic acid sodium salt), is a dual (mixed) ERA that was developed by (Glaxo) SmithKline (GSK; Ref. 7; reviewed in Ref. 70). Even though this compound presents a high affinity to the ETA receptor subtype, it also binds to ETB-Rs and therefore cannot be considered a selective ERA. Given the effects of ETs to elicit prolonged vasoconstriction and to enhance cell proliferation and stimulation of extracellular matrix accumulation, and following encouraging preclinical studies using animal models of hypertension and left ventricular hypertrophy (reducing blood pressure [BP], preventing cardiac hypertrophy, and preserving myocardial function), GlaxoSmithKline was swift to move forward with clinical studies. Enrasentan has been tested in clinical studies as treatment of patients with heart failure (New York Heart Association [NYHA] Class 2–3) (see below).
Tezosentan.
This mixed (dual) ERA-A/B was developed by Roche and then licensed to Actelion: Ro 61-0612 ([5-isopropyl-pyridine-2-sulfonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(2–1H-tetrazol-5-yl-pyridin-4-yl)-pyrimidin-4-ylamide sodium salt, 1:2]). It is an intravenously active ERA with a short half-life (71). It was reported to alter coronary hemodynamics and oxygen metabolism during exercise in dogs (72). It successfully decreased serum creatinine, increased glomerular filtration rate, and maintained renal architecture in kidneys after ischemia (73). Tezosentan was selected for trials in patients with acute heart failure (74, 75).
Selective ERA-A (In Alphabetical Order).
Ambrisentan.
LU 208075 was first developed by Knoll GmbH and subsequently acquired by Abbott. Later, this compound was licensed to Myogen, who are developing it as a treatment for PAH (76). LU 208075 is an orally active, propanoic acid class, nonpeptide selective ETA receptor antagonist (ERA-A) that was initially tested on the contraction and relaxation of isolated basilar arteries (77). One of its key advantages is an excellent pharmacokinetic profile (9–15 hrs of circulating half-life, allowing once a day dosing). Though very potent, it has relatively weaker selectivity (260×) for the ETA-R subtype (compared to other ERA-As; see Table 1). The window of dosing may constitute an important aspect. Although ARIES-2 study demonstrated a well-defined dose-response between 2.5 and 5 mg. Ambrisentan was reported to improve hepatic warm ischemia/reperfusion injury in pigs (78) and rats (79), but failed to improve survival and pancreatic damage during acute experimental pancreatitis (80). More recently, Actelion designed a novel class of ERA-A based on a 1,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepin-2-one scaffold of ambrisentan (81). Another structurally-related ERA-A from Knoll (LU 302146) has been tested in experimental models of uremic cardiomyopathy and transplant vasculopathy (82–84).
Atrasentan.
ABT-627 (A-147627; [2-(4-methoxy-phenyl)-4-(1,3-benzodioxol-5-yl)-1-(N,N-di(n-butyl)amino carbonylmethyl)-pyrrolidine-3-carboxylic acid]; trade name, Xinlay) was developed by Abbott (26, 27, 85). It is an orally available, nonpeptidic, highly selective ERA-A that was chosen for clinical development, as reviewed by Norman in 2002 (86). Quite early on, Nelson et al. (87) studied the role of the ET system in advanced and metastatic prostate cancer. This led to the present interest by Abbott in the pathological role of ETs in modulating mitogenesis, angiogenesis, and apoptosis in various forms of cancer (88, 89).
Avosentan.
SPP301 (Ro 67–0565: 5-methyl-pyridine-2-sulfonic acid [6-methoxy-5-(2-methoxy-phenoxy)-2-(pyridine-4-yl)-pyrimidin-4-yl]-amide is, according to Speedel, which licensed it from Roche, an orally available and competitive ERA-A. However, it demonstrates relatively weaker selectivity for the ETA subtype (50–600×).1 Even though we grouped it with the other selective ERA-A (see Table 1), it may ultimately be classified as a dual ERA. Its hydroxymethyl metabolite is Ro 68–5925. The pharmacokinetic and pharmacodynamic profile of SPP301 as well as the tolerability in healthy subjects has been described (21, 90). SPP301 is now in Phase III clinical development for diabetic nephropathy (see below).
Clazosentan.
Ro 61-1790 ([5-methyl-pyridine-2-sulfonic acid 6-(2-hydroxy-ethoxy)-5-(2-methoxy-phenoxy)-2-(2–1H-tetrazol-5-yl-+++pyridin-4-yl)-pyrimidin-4-ylamide]) is a nonpeptidic, hydrosoluble, competitive and selective ERA-A developed by Roche (29) and then licensed by Axovan (Switzerland), which was acquired thereafter by Actelion. Clazosentan was specifically designed and selected for its efficacy in models of cerebral vasoconstriction. It was developed for parenteral use (iv) for preventing delayed cerebral vasopasm in patients with subarachnoid hemorrhage. Clinical studies have been conducted to assess its effects on cardiac output, aortic pressure, and pulse wave velocity (91) as well as renal function (92).
Darusentan.
LU 135252 ((+)-(S)-2-(4,6-dimethoxy-pyrimidin-2-yloxy)-3-methoxy-3,3-diphenyl-propionic acid (32) is another compound developed by Knoll, acquired by Abbott, and licensed to Myogen. It is marketed as an orally available, selective ERA-A (see Table 1), thus still presenting a relatively high affinity for the ETB subtype (30, 31) (Table 1). Like ambrisentan, darusentan has a long half-life (16–18 hrs). It has completed Phase II-b clinical development for the treatment of resistant systolic hypertension (Tables 2 and 5). The efficacy of darusentan was tested in several experimental animal models of diseases, suggesting other potential therapeutic uses.
Edonentan.
Early work with the related compound, BMS-193884 (N-(3,4-dimethyl-5-isoxazolyl)-4′-(2-oxazolyl)[1,1′-biphenyl]-2-sulfonamide), revealed its high selectivity for the ERA-A (>13,000-fold with the constant of inhibition (Ki) = 1.4 nM for ETA and 18.7 μM for ETB; Ref. 28). An optimum pharmacologic profile contributed to its selection as a clinical candidate for studies in congestive heart failure (CHF; Tables 1, 2, and 5) following promising results from preclinical studies in rodent and porcine models (108–110). This compound was also tested in models of monocrotaline-induced PAH. It was replaced by edonentan (BMS-207940; (N-[[2′-[[(4,5-dimethyl-3-isoxazolyl)amino]sulfonyl]-4-(2-oxazolyl)[1,1′-biphenyl]-2-yl]methyl]-N,3,3 trimethylbutanamide), the backup compound, an even more potent and selective ERA-A (80,000-fold with Ki = 0.010 nM for ETA-R) (33).
Sitaxsentan.
TBC11251 (N-(4-chloro-3-methyl-5-isoxazolyl)-2-[(6-methyl-1,3-benzodioxol-5-yl) acetyl]-3-thiophenesulphonamide) is a compound under clinical development by Encysive Pharmaceuticals (formerly Texas Biotechnology Corporation). Sitaxsentan has been reported to be an orally available (50%–60% and 90%–100% bioavailability in rat and dog, respectively) and highly selective ERA-A (6500-fold with Ki of 0.35 nM for ETA; 34). In addition, sitaxsentan exhibits approximately 90% oral bioavailability in human subjects and has a t½ of 10 hrs in patients with PAH. Thus far, 31 clinical studies have been conducted with sitaxsentan, including three major randomized, placebo-controlled trials (Sitaxsentan to Relieve Impaired Exercise [STRIDE] trials; see below) in patients with PAH.
S-0139 (SB-737004).
S-0139 (27-O-3-[2-(3-carboxy-acryloylamino)-5-hydroxyphenyl]-acryloyloxy myricerone, sodium salt; also known as SB-737004) is a highly selective nonpeptidic ETA-R antagonist (Ki: 1 nM on ETA-R vs. 1000 nM on ETB-R) developed by Shionogi (Japan). The compound has a 1000-fold more potency for ETA as compared to ETB receptors (Table 1; Ref. 188). A joint venture was finalized with GSK in 2001-Q4 to codevelop and commercialize four compounds contributed by Shionogi and one by GSK, including S-0139 for the potential treatment of hemorrhagic and ischemic stroke (187, 189).
Preclinical experimental data with S-0139 suggested its potential beneficial effects on brain edema and injury in rats, CHF in mice, and renal cortical blood flow reduction in rats (190–192).
TBC 3711.
TBC 3711 (N-(2-acetyl-4,6-dimethylphenyl)-3-(3,4-dimethylisoxazol-5-ylsulfamoyl)thiophene-2-carboxamide) is another of Encysive’s orally available, nonpeptidic, selective ERA-A (35). It has an oral bioavailability of approximately 100% in rats, high potency (mean inhibitory concentration [IC50] on ETA-R = 0.08 nM), and an ETA selectivity of >100,000-fold (Table 1). It has completed a Phase I clinical trial and was well tolerated with reported pharmacokinetics in humans (t½ = 6–7 hrs, oral availability > 80%). In a preclinical experimental study, TBC3711 (22 mg/kg/day) has been reported to reduce neonatal hypoxia-induced pulmonary hypertension in 1-day-old piglets (112).
YM598.
Yamanouchi (now called Astellas Pharma after its merger with Fujisawa) developed ethenesulfonamide derivatives as a potent, orally active and selective type of ERA-A (36, 37) through the modification of bosentan. YM598 ((E)-N-[6-methoxy-5-(2-methoxyphenoxy)[2,2′-bi-pyrimidin]-4-yl]-2-phenylethenesulfonamide monopotassium salt; Fig. 1) is the lead compound and is 816-fold more selective for the ETA receptor (Ki = 0.697 and 569 nM for ETA and ETB respectively; Table 1; Refs. 38–40). The first indication selected for YM598 was prostate cancer (as with Abbott and AstraZeneca), as suggested by preclinical data in mice (113, 114). YM598 was also studied and proven effective in animal models of chronic hypoxia-induced PAH, monocrotaline-induced PAH, hypertension (normal and low-renin), myocardial infarction-induced CHF, postischemic left-side CHF, and type 2 diabetic nephropathy.
ZD4054.
AstraZeneca has been working in the field since 1994 and developed ZD1611 and ZD2574 (115). More recently, they have developed an orally active, more potent and selective ERA-A, ZD4054 (5-(dimethylamino)-N-(5chloro-3-methoxy-2-pyrazinyl)-1-naphthalenesulfonamide; Refs. 41, 108). This novel antagonist emerged from structure activity relationship (SAR) studies conducted with BMS-182874 (41). Because ETA-R activation by ET-1 mediates events that regulate mitogenesis, apoptosis, angiogenesis, and metastasis in tumors, AstraZeneca has entered this area as well as Abbott. AstraZeneca completed a Phase II-a trial (see below; Tables 2 and 5) and initiated a Phase II-b trial in 250 patients with prostate cancer (see below; Tables 2 and 7).
Selective ERA-Bs.
Even though no trials have involved the use of selective ERA-Bs, they merit mention because highly potent, nonpeptidic, selective compounds have been developed and tested in numerous preclinical experimental animal models of disease, helping us to better understand the physiopathology of the ET system.
A-192621.
Abbott’s selective ERA-B (1400-fold selectivity; ETA Ki = 6.5 μM vs. ETB Ki = 4.5 nM; [2R-(2alpha,3beta,4alpha)]-4-(1,3-benzodioxol-5-yl)-1-[2-[2,6-(diethylphenyl)amino]-2-oxoethyl]-2-(4-propoxy-phenyl)-3-pyrrolidine carboxylic acid; 193) has been widely used (70 publications since 1999). This pharmacologic tool enabled us to clearly identify physiological roles for ETB-mediated responses, including the regulation of basal BP in normal, conscious mice (194) and primates/cynomolgus monkeys (195).
Approved New Drug Application (NDA): The Homologation of a New Class of Drug Through Clinical Trials
Actelion, with its bosentan, became the first to successfully emerge from the U.S. Food and Drug Administration (FDA)’s NDA homologation in 2001. Endothelin receptor antagonists became a new class of drug bearing the “-sentan” suffix.
Formally Completed and Ongoing Clinical Academic Studies and Trials in Control Subjects and Patients
Below, we briefly discuss clinical academic studies, as well as small-scale clinical trials, in healthy subjects and patients, that were conducted using the first generations of ERAs (peptidic, like BQ-123, and some others that are nonpeptidic). Such studies, many of which are ongoing and recruiting patients, are supported by public hospitals and/or government agencies in order to increase understanding of the pathophysiology and mechanisms of action of the ET system (Table 4).
Full-length clinical trials are mostly supported by biopharmaceutical industries. We have summarized them by separating between completed (Tables 4 and 5) and ongoing (Tables 6 and 7) clinical trials, while reviewing them one by one, elaborating on each trial’s name, phase of development, sponsor, target, study design, duration, formulation, route of administration, and, if completed, results, supported by key references when available.
Infusion of Exogenous ET-1 and Related Agonists of the ET System.
Initial studies, performed to elucidate the role of the then-recently discovered ET-1, involved the infusion of the exogenous peptide (agonist at both ETA and ETB receptors) and/or sarafotoxin-6c (S6c; an ETB receptor agonist) into the forearm circulation of healthy volunteers. These studies confirmed that, as in earlier animal experiments (141), ET-1 acts as a long-lasting, potent vasoconstrictor in man (116). Later, it was shown that ET-1 exerts a biphasic effect: low-dose ET-1 caused vasodilatation, whereas higher doses resulted in vasoconstriction (117). This biphasic response was also seen when bolus doses of S6c were infused into healthy subjects (118), suggesting that activation of ETB receptors might cause vasodilatation as well as vasoconstriction. When infused systemically, ET-1 exerted a marked pressor effect associated with a reduction in renal blood flow and decreased natriuresis (119). Such changes were not seen following systemic infusion of ET-3 (an ETB receptor agonist), indicating that these systemic and renal vasoconstrictor effects of ET-1 in healthy volunteers are mediated predominantly by ETA receptors.
Infusion of ERAs.
Since their development by Banyu (Merck), appropriate doses of the ERA-A BQ-123 (1) and the ERA-B BQ-788 (2) have been used as highly selective probes to determine the physiological role of ET-1 both in health and in a range of cardiovascular, high-risk disease states. Nevertheless, BQ-123 and BQ-788 have been shown to exert nonselective ET blocking effects at higher doses (142, 123). Recently, such studies have been extended to investigate the potential therapeutic role of these ERAs in reversing the endothelial dysfunction seen in cardiovascular disease.
Physiological Role of ET-1.
When first used in humans, BQ-123 infusion into the forearm circulation of healthy volunteers caused marked vasodilatation, strongly suggesting that endogenous ET-1 maintains vascular tone through basal activation of ETA receptors (120). In contrast, local infusion of BQ-788 resulted in vasoconstriction, indicating that ETB receptors act to reduce basal vascular tone (121). The vasodilatory action of BQ-123 was significantly reduced by ETB blockade and almost abolished when nitric oxide (NO) synthesis was inhibited (121). This indicated that it is attributable, to not only direct blockade of ET-1 binding to vasoconstrictive ETA receptors on vascular smooth muscle, but also enhanced endogenous NO generation and the preservation of ETB receptor-mediated vasodilator tone. In contrast, the response to BQ-123 is not altered following cyclooxygenase inhibition with aspirin, providing strong evidence that this response is not dependent upon endothelium-derived prostaglandins.
Systemic infusion of BQ-788 into healthy volunteers resulted in a significant rise in peripheral vascular resistance, although, because of a compensatory fall in heart rate, no change in BP was seen (122). Within the vasculature, ETB receptors are expressed on both vascular smooth muscle cells, where they cause vasoconstriction, and on the endothelium, where their activation results in vasodilatation. Along with the forearm studies detailed above (121), this suggests that the net effect of ETB activation in healthy subjects favors vasodilatation via the endothelial ETB receptors. This study also investigated the effect of systemic ETB antagonism on plasma ET-1 concentrations. As expected, and as observed in preclinical studies with BQ-788, ETB selective antagonism leads to a 5- to 7-fold increase in ET-1 plasma concentrations in the rat, the administration of an ERA-B, which impairs clearance of ET-1 (143), resulted in an increase in the plasma concentration of ET-1. In contrast, when BQ-123 was infused systemically over a wide range of doses, no difference in plasma ET-1 was found (123). Here, BQ-123 dose-dependently decreased systemic vascular resistance and mean BP (123), providing further evidence that endogenous ETA activation contributes to the maintenance of basal systemic vascular tone.
Role of ET-1 in Cardiovascular Disease.
Having used BQ-123 and BQ-788 to help reveal the physiological roles of ET-1, investigators were swift to perform similar experiments in patients with either risk factors for, or established, atherosclerotic disease, in order to determine whether the ET system is active in such pathological states.
BQ-123 infused into the forearms of CHF patients resulted in an increase in forearm blood
flow (124). Two independent groups then showed that local
infusions of BQ-123 and BQ-788 (125) or of the mixed ERA, TAK-004
(126), causes a greater vasodilatation in patients with
essential hypertension than in healthy subjects. Moreover, vasoconstriction to
NG-monomethyl-
In hypercholesterolemic patients, BQ-123 infusion also increased forearm blood flow compared to healthy controls. However, this was reversed by coinfusion with the ERA-B, BQ-788 (127). A similar result was seen in an equivalent study in Type 2 diabetic patients (128).
Studies have also been performed to investigate the effect of ERAs in patients with established atherosclerosis (defined in this study as the combination of intermittent claudication with coronary artery disease; Ref. 131). As expected, BQ-123 caused an increase in forearm blood flow in both groups. However, BQ-123 and BQ-788 infused together had a negligible effect on vascular tone in control subjects. In contrast, combined ERA blockade caused a greater dilatation in atherosclerotics than that seen with BQ-123 alone. This suggests a greater functional significance of vasoconstrictive vascular smooth muscle ETB than dilatory endothelial ETB in these patients.
The important role of ETA-dependent vascular tone in the coronary circulation was demonstrated by the vasodilatory effect of intracoronary infusions of BQ-123 (132). It was found to be exaggerated in subjects with known coronary artery disease (133, 137). Intracoronary BQ-788 caused microvascular constriction, partly as a result of reduced ET-1 clearance and reduction in NO production (134).
As well as being used in forearm and coronary circulation studies, ERAs have also been used following systemic administration. In patients suffering from hypertensive chronic renal failure, BQ-123 substantially increased renal blood flow and reduced renal vascular resistance. These effects were not seen with mixed ET blockade (135). In contrast, infusion of BQ-788 resulted in both renal and systemic vasoconstriction. Similarly, infusion of BQ-123 reduced both systemic and pulmonary vascular resistance in CHF patients to a greater degree than following coinfusion of both BQ-123 and BQ-788 (136). BQ-788 alone worsened hemodynamic variables and had deleterious effects on a number of patients involved in the study.
ERA Reverse Endothelial Dysfunction.
The demonstration that ERA-As cause vasodilatation, not only through competitive inhibition of ET-1 binding to ETA receptors, but also through increasing the generation of endogenous NO (121), encouraged many groups to study the effect of ERAs on endothelial function more directly.
The dilatation of small arteries in response to acetylcholine is dependent upon endothelial-derived NO, and has been taken as a measure of endothelial function. Intra-coronary infusion of BQ-123 enhanced acetylcholine-induced vasodilatation of the coronary arteries of patients with atherosclerosis (137) to a greater extent to that seen following coinfusion of BQ-788 and BQ-123 (134). Neither compound altered the endothelium-independent vasodilatation to sodium nitroprusside. In the forearms of both hypertensive patients (138) and patients with atherosclerosis (139), mixed blockade with BQ-123 and BQ-788 similarly potentiated the response to acetylcholine without affecting the response seen to sodium nitroprusside. Mental stress–induced impairment of endothelial function, brought on in healthy subjects by a 3-min mental stress task, could also be partially reversed by infusion with BQ-123 (140).
Clinical studies with the Banyu peptide ERAs, as probes for ETA and ETB receptor blockade, have allowed a much fuller understanding of the ET system in health and disease as well as of its role in modifying endothelial function. Such knowledge has informed the choice of targets for subsequent clinical trials.
Completed Clinical Trials in Control Subjects and Patients
Completed Clinical Trials with Dual ERA (cf. Tables 2 and 4).
BREATH-1.
The Bosentan Randomized Trial of Endothelin Antagonist Therapy (BREATH-1) was the first large trial involving an ERA, fully sponsored by Actelion, to investigate the effects of an ERA in patients with iPAH and PAH associated with connective tissue diseases (such as scleroderma). It became the first-in-class ERA. Both Phase II-b and Phase III studies were successful in supporting the hypothesis that the ET system is a key component in severely compromised patients with PAH (45–47). The beneficial effects of bosentan on exercise capacity were maintained for at least 20 weeks. Furthermore, bosentan led to a significantly greater improvement in secondary efficacy endpoints such as the Borg dyspnea index, World Health Organization (WHO) functional class, and cardiopulmonary hemodynamic parameters (such as cardiac index [CI], pulmonary vascular resistance [PVR], pulmonary artery pressure [PAP], pulmonary capillary wedge pressure [PCWP], and mean right atrial pressure [RAP]) compared with placebo. Bosentan significantly reduced the incidence, and delayed the onset, of clinical worsening of PAH compared with placebo. Adverse events occurred with similar or greater frequency with bosentan (125 mg twice a day [bid]) compared to placebo, including headache, syncope, flushing, and abnormal hepatic function (see section below). Those side effects that occurred less frequently with bosentan (125 mg bid) than with placebo included dizziness, worsening of symptoms of PAH, cough, and dyspnea. A BREATH-1 substudy in systemic sclerosis (SSc; scleroderma)-associated PAH patients also demonstrated improved outcome with bosentan treatment, preventing clinical deterioration (Tables 2 and 4).
BREATH-4 with Bosentan.
This was a small (n = 16), prospective, noncomparative cohort study investigating the effects of bosentan in human immunodeficiency virus (HIV)-associated PAH patients (49). Treatment resulted in improvements in the endpoints of exercise capacity, cardiopulmonary hemodynamics, functional class, and quality of life (Tables 2 and 4; Ref. 49). During the study, there were no deaths, and no patients required epoprostenol treatment. Hepatic tolerability was similar to that reported in patients with iPAH. Bosentan treatment did not worsen either CD4 count or HIV viral load. During 12 months of follow-up in 12 patients, during which bosentan treatment was continued, no deterioration in functional class was seen. Despite the co-administration of potentially hepatotoxic antiretroviral therapies, the safety profile of bosentan was good and no clinically relevant drug interactions were observed (49).
Comparing Bosentan with Prostacyclin Treatment.
This study compared the survival of patients with class III iPAH treated with first-line oral bosentan versus the historical cohort of patients started on iv epoprostenol (50). The use of bosentan delayed the initiation of epoprostenol. Kaplan-Meier survival estimates were 97% and 91%, after 1 and 2 years, respectively, in the bosentan cohort, and 91% and 84% in the epoprostenol cohort. When matched cohorts of 83 patients each were selected, survival estimates were similar. In the bosentan cohort, 87% and 75% of patients followed for 1 and 2 years remained on monotherapy. Therefore, no evidence was found to suggest that initial therapy with oral bosentan, followed by other treatment if needed, adversely affected long-term outcome compared with initial intravenous epoprostenol in class III iPAH patients (50).
ENABLE.
In 1998, oral bosentan was used in a double-blind and randomized short-term trial as an add-on therapy in patients (n =24 +12 placebo) with symptomatic severe CHF conventionally treated with diuretics, digoxin, and angiotensin-converting enzyme inhibitor (ACEi) over 2 weeks (143). On Day 1, bosentan significantly decreased mean arterial BP, PAP, PCWP and RAP. Cardiac output (CO) increased, whereas heart rate (HR) remained unchanged. After 2 weeks of bosentan therapy, CO further increased, whereas systemic vascular resistance (SVR) and PVR fell (144). Following this encouraging short-term hemodynamic effect of oral therapy with bosentan in heart failure patients who were symptomatic with standard triple-drug therapy, Endothelin Antagonist Bosentan for Lowering Cardiac Events in Heart Failure (ENABLE) was swiftly organized.
In this large trial, 1613 patients with severe heart failure (left ventricular ejection fraction <35%, NYHA class III-b–IV) were randomized to receive either bosentan (125 mg bid after a 4-week titration phase at 62.5 mg bid) or placebo. The preliminary results were presented at the 51st Annual Scientific Session of the American College of Cardiology (March 2002, Atlanta, Georgia) and did not demonstrate a benefit with bosentan treatment. The primary endpoint of all-cause mortality or hospitalization for heart failure was reached in 321 of 808 patients on placebo, and in 312 of 805 patients receiving bosentan. Treatment with bosentan appeared to confer an early risk of worsening heart failure necessitating hospitalization, as a consequence of fluid retention. It has been suggested that further studies using even lower doses of bosentan or more aggressive concomitant diuretic therapy may avoid this adverse effect. Such results from the ENABLE study created doubt about the potential benefits of ERA in the treatment of CHF (51).
REACH-1 with Bosentan.
In the Research on Endothelin Antagonism in Chronic Heart Failure (REACH) study (52), which preceded the ENABLE study, the long-term effects of bosentan (target dose 500 mg bid; n = 244) versus placebo (n =126) in patients with CHF (NYHA class IIIB/IV) were assessed. This trial was halted prematurely because of increased incidence of elevated liver transaminase levels. When the trial was stopped, however, patients who had been maintained on therapy over a 6-month period demonstrated a trend toward reduced risk of heart failure-related mortality and morbidity (52).
ENCOR.
In the Enrasentan Cooperative Randomized Evaluation (ENCOR), treatment of 419 patients with CHF (Class II–III) randomized to either the dual ERA, enrasentan, or placebo failed to show benefit in a composite end point including NYHA class, hospitalization rate, and global assessment. In fact, the study showed a trend in favor of placebo: there was a significantly greater likelihood of being hospitalized for heart failure—almost 3-fold—for patients randomized to enrasentan (53).
RITZ-1.
There were four Randomized Intravenous Tezosentan (RITZ) studies in patients with acute decompensated heart failure (ADHF). Phase I investigated the tolerability, pharmacokinetics, and pharmacodynamics during chronic infusions of tezosentan in healthy male subjects (145, 54). Both doses tested (Table 4) were well tolerated with headaches being the most frequently reported adverse event (incidence of 75%–100% for tezosentan and 50% for placebo). Plasma concentrations of tezosentan rapidly approached steady state and did not change upon prolonged infusion. A two-compartment model described its pharmacokinetic profile. The half-lives of the two disposition phases were approximately 0.10 and 3.2 hrs. Circulating plasma concentrations of ET-1 increased rapidly during infusion, compared with predose values, and did not change during the 72-hr infusion. The volume of distribution at steady state (approximately 16 liters) and the clearance (approximately 30 liters/hr) were considered independent of dose, in view of the wide dose range explored. Additional doses (5, 20, 50, 100, 200, 400, and 600 mg/hr) were also infused for 1 hr (55). No additional clinically relevant changes in vital signs or in electrocardiographic or clinical laboratory parameters occurred.
A second safety trial was conducted with the co-administration of cyclosporine and tezosentan. Cyclosporine caused a 4-fold increase in the exposure to tezosentan (56). All subjects on the combined regimen reported headache, hot flushes, and nausea/vomiting. Some of these symptoms were of severe intensity. The symptoms did not correlate with the circulating plasma concentrations of tezosentan.
In a third safety study, no notable differences in safety and tolerability variables were detected between tezosentan-treated and placebo-treated patients when infused over 4 to 6 hrs.
A fourth pilot safety trial was conducted in patients with advanced heart failure infused over 48 hrs (57). The safety and tolerability, as well as the hemodynamic stability, following tezosentan treatment revealed no episodes of hypotension requiring withdrawal of therapy occurred. Hemodynamic rebound was not observed after abrupt cessation of the infusion. In addition, there were no reports of worsening heart failure in tezosentan-treated patients up to 28 days following the infusion. The most common side effect observed during the infusion was headache. Echocardiographic Doppler measurements suggested improvements in CI, PCWP, and relaxation properties as well as in diastolic and systolic function. Thus, tezosentan was well tolerated with no new safety concerns emerging (57).
RITZ-2.
This study was designed to determine the effective dosage range, hemodynamic effects, and tolerability of tezosentan in patients with advanced NYHA class III heart failure (58, 59). It acutely improved hemodynamic parameters (significant increase in CI and decreases in PVR and SVR without changes in heart rate). However, a consistent greater decrease in right-sided pressures and mean systemic BP did not reach statistical significance. Hemodynamic changes were dose-dependent with maximal effects at 20 and 50 mg per hour. Tezosentan was well tolerated. Despite an increase in circulating plasma concentrations of ET-1, hemodynamic rebound was not observed. Thus, the favorable effects on CI and vascular resistance, without changes in heart rate, suggested that tezosentan could be beneficial in the treatment of ADHF (58).
RITZ-3.
This was a further Phase II-b trial intended to enroll more patients (NYHA class III–IV) and study them over a longer duration (from 1 [Ref. 58] to 6 hrs follow-up) in combination with a diuretic (60). However, it was deemed unnecessary from a regulatory point of view by Actelion and was never conducted.
RITZ-4.
Thus far, tezosentan, as a dual ERA, was shown to improve CI/CO and reduce PVR and SVR in initial human acute decompensated heart failure studies (see above). The next step included a dose-optimization trial (design in Refs. 146, 147) as part of Phase III studies that would provide valuable data regarding the efficacy and tolerability benefits, as well as the morbidity and mortality. However, following disappointing trial results, a new 2-year Phase III study in ADHF was planned using lower doses of the compound (75). This additional Phase III trial was designed as a multicenter, randomized, double-blind, placebo-controlled trial with 193 enrolled patients with ADHF and acute coronary syndromes. No significant differences were observed between placebo and 50 mg/hr tezosentan infusion in the composite primary end point (e.g., death, worsening of heart failure, recurrent ischemia, and recurrent or new myocardial infarction within 72 hrs after the initiation of the treatment with tezosentan). Symptomatic hypotension was more frequent in the treatment group. Thus, at the doses studied, tezosentan did not result in a significant improvement (61).
Study 205 with Tezosentan from Actelion.
This study was a Phase II trial designed to optimize tezosentan dosing in patients with ADHF who required right heart pressure monitoring. The study compared the effects of iv tezosentan (0.2, 1, 5, and 25 mg/hr) versus placebo on CI, PCWP, urine output, and serum creatinine. The three highest doses showed significant increases in CI and decreases in PCWP. However, the maximum effect was evident only at 5 mg/hr and the highest dose decreased urine output.
VERITAS-1 and -2.
RITZ studies left us uncertain whether ERA could be effective in patients with ADHF. Morbidity and mortality had not been evaluated. The Value of Endothelin Receptor Inhibition with Tezosentan in Acute Heart Failure Study (VERITAS) consisted of two identical double-blind, randomized, placebo-controlled, multicenter trials, designed to enroll at least 1760 patients (Table 4) and study the effect of tezosentan on mortality and rate of clinical worsening, The program was discontinued in November 2005 because of the low probability of achieving a significant treatment effect over the first 24 hrs of treatment and at 7 days (62).
Avosentan by Speedel.
Based on a pilot study, Phase I through II-b trials have been completed. 286 patients were enrolled in a randomized, placebo-controlled, double-blind, parallel-design dose-range study (see Table 4; Ref. 44). The study compared the effects of a 12-week therapy with SPP301 (5, 10, 25, 50 mg) or placebo, in addition to standard treatment on urinary albumin excretion rate. SPP301 decreased urinary albumin excretion rate significantly, whereas total cholesterol also decreased significantly. Creatinine clearance and BP were unaffected. The main adverse effects were peripheral edema and headache. Because it was administered concomitantly with ACEi, SPP301 presents additional benefits toward treating this disease.
Completed Clinical Trials with Selective ERA-A (See Tables 2 and 5).
Theoretically, selective antagonism of the ETA, rather than dual ET antagonism, could block the deleterious vasoconstrictive and promitogenic ETA-mediated effects without altering ETB-mediated clearance and vasodilation actions (though NO and prostacyclin release).
AMB-220 with Ambrisentan.
The purpose of this Phase II study was to examine the efficacy and safety of four doses of ambrisentan in patients with iPAH of various etiologies (associated with collagen vascular disease, anorexigen use, or HIV). This double-blind, dose-ranging (four doses) study revealed an improvement for all primary efficacy end points: 6-min walk distance (6MWD; +36.1 m, P < 0.0001), Borg dyspnea index, WHO functional class, and cardiopulmonary hemodynamics, such as mean PAP (−5.2 mm Hg, P < 0.0001) and CI (+0.33 liters min−1 m−2 P < 0.0008). Adverse events, such as elevated liver function tests, were mild and unrelated to the dose, but peripheral edema reached 25% (see section below; Table 5; Ref. 93).
A long-term study of subjects who participated in AMB-220 has demonstrated durable efficacy and safety for more than two years.
ARIES-2 with Ambrisentan.
This was a Phase III, randomized, double-blind, placebo-controlled, multicenter, efficacy and safety studies in subjects with PAH (Table 7). This study evaluated two doses of ambrisentan 2.5 and 5 mg per day in 186 PAH subjects (62 subjects/arm). The primary efficacy endpoint was the change from baseline in the 6MWD evaluated after 12 weeks of therapy compared to placebo. Secondary endpoints include Borg dyspnea index, WHO functional class, the SF-36 health survey, and time to clinical worsening. Results of the trial demonstrated that both doses of ambrisentan significantly improved the placebo-corrected mean 6MWD (32.3 m and 59.4 m for the 2.5 mg and 5 mg dose groups, respectively). Significant improvements in time to clinical worsening compared to placebo were also observed for both doses (Table 5). The most frequent adverse event was headache, which occurred in 12.7% of patients in the 5 mg dose group and 7.8% in the 2.5 mg dose group, compared to 6.2% in the placebo group. No patients treated with ambrisentan developed serum aminotransferase concentrations greater than three-times the upper limit of the normal range and ambrisentan had no apparent effect on the activity or dosage of warfarin-type anticoagulants.
ARIES-1, a companion study to ARIES-2, will examine doses of 5 mg and 10 mg of ambrisentan compared to placebo. ARIES-E, a long-term study of patients who participated in ARIES-1 and ARIES-2 currently has more than 325 patients being treated for periods up to two years.
BMS-193884.
A pilot study was conducted with BMS-193884, a selective ERA-A for the treatment of mild to moderate erectile dysfunction (102). Human volunteer subjects (n = 53) were selected to assess the safety and efficacy of BMS-193884. Though the drug was well tolerated (oral 100 mg), it did not improve erectile function significantly during office visits or home use when compared to placebo.
The same compound was also assessed in a Phase I trial in November 1996 and continued into Phase II trials in patients with CHF (148). No reports have emerged since then because it was probably discontinued.
Edonentan.
BMS-207940, a second-generation analog of BMS-193884, entered Phase I clinical trials by April 2002. Filing for NDA was expected to take place in 2004, aimed at CHF (148). No news has emerged from these trials and the program was seemingly dropped (the Bristol-Myers Squibb website does not reveal any information).
EARTH with Darusentan.
The Endothelin-A Receptor Antagonist Trial in Heart Failure (EARTH) study aimed at measuring the effects of long-term ET blockade on left-ventricular (LV) remodeling and clinical outcomes in patients with CHF (Table 5). In this study, 642 patients with NYHA Class II–IV CHF (receiving either an ACEi, beta blocker or an aldosterone antagonist) were randomized to treatment with darusentan or placebo over 24 weeks (99). The primary endpoint was the change in left ventricular end systolic volume (LVESV) measured by magnetic resonance imaging. No significant difference was seen in left ventricular end systolic volume (assessed in 485 (76%) paired magnetic resonance imaging data at baseline and 6 months) from that following placebo treatment for any dose of darusentan. Furthermore, there was no difference seen in terms of mortality or the progression of CHF, even though heart failure worsened in 71 (11.1%) patients, and 30 (4.7%) died during the study. Circulating plasma concentrations of ET-1 increased dose-dependently in all groups receiving darusentan. Darusentan did not improve cardiac remodeling (Table 5).
HEAT-CHF with Darusentan.
Darusentan is a potent and selective ERA-A (see Table 1; Ref. 31). The Heart Failure ETA Receptor Blockade Trial (HEAT) investigated hemodynamic and neurohumoral effects of 3 weeks of treatment with various dosages (30, 100, or 300 mg/day) of the orally available darusentan in addition to standard therapy in patients with CHF (100). A total of 157 patients with CHF (NYHA class III of at least 3 months duration), PCWP ≥ 12 mm Hg, and cardiac index (CI) ≤ 2.6 liters/min/m2 were randomly assigned to double-blind treatment with placebo or darusentan in addition to standard therapy. A 21-day administration period of darusentan increased the CI, but this did not reach statistical significance compared with placebo. The increase in CI was significantly more pronounced after 3 weeks of treatment (P < 0.0001 versus placebo). PCWP, PAP, PVR, and RAP remained unchanged. Heart rate, mean BP, and plasma catecholamines remained unaltered. However, SVR decreased significantly. Higher dosages were associated with a trend to more adverse events (including death), particularly early exacerbation of CHF without further benefit to hemodynamics compared with moderate dosages.
HEAT-HTN with Darusentan.
The Hypertension Endothelin Antagonist Treatment (HEAT) study was a randomized, double-blind, placebo-controlled, multicenter, parallel-group study that investigated the antihypertensive efficacy and safety of darusentan in subjects with moderate essential hypertension (Tables 2 and 5). A total of 392 subjects were randomized to placebo or one of three doses of darusentan (10, 30, or 100 mg) for 6 weeks. The study results demonstrated a statistically significant and dose-dependent decrease in systolic and diastolic BP as compared to placebo. The mean, placebo-corrected change from baseline in systolic BP was −6.0 mm Hg on 10 mg, −7.3 mm Hg on 30 mg, and −11.3 mm Hg on 100 mg darusentan, following the 6-week treatment period. Significant placebo-corrected reductions in diastolic BP were also observed (−3.7, −4.9, and −8.3 mm Hg, respectively). Darusentan was well tolerated in this subject population.
DAR-201 with Darusentan.
DAR-201 was a Phase II-b randomized, double-blind, placebo-controlled study designed to examine the safety and efficacy of increasing doses of darusentan in patients with resistant systolic hypertension (Tables 2 and 5). A total of 115 subjects with resistant systolic hypertension who were receiving combination therapy with three or more antihypertensive drugs (including a diuretic) at documented full doses were randomized to darusentan or placebo for 10 weeks. The maximal dose (300 mg/day) of darusentan was associated with a statistically significant, placebo-corrected reduction in systolic and disatolic BP of 11.6 and 7.0 mm Hg. Darusentan was generally well tolerated. There were no serum aminotransferase elevations above two times the upper limit of normal.
Studies with Atrasentan.
Phase I trials (I-a, single-dose and I-b, multiple-dose) were conducted in 182 (seven substudies) and 234 (six substudies) healthy subjects, respectively. These studies were accompanied by drug-interactions studies (four substudies, total 52 subjects), an open-label oncology study (four substudies, total 111 patients) and continuation in 47 patients (three open-label substudies).
M96-500, M96-594, and M00-211 with Atrasentan.
Atrasentan was granted Fast Track review status following an initial Phase I/II pivotal trial in 46 patients (M96-499, M97-661 and M02-531), allowing for a rolling NDA in metastatic hormone-refractory prostate cancer (HRPC; Tables 2 and 5; Ref. 86). Abbott is completing the NDA ahead of the 2005 timetable predicted. The M96-500 and M96-594 Phase II trial data were disclosed in February 2003. These showed a trend toward a delay in time to disease progression, though the trend failed to reach statistical significance (95). A meta-analysis of recently pooled data combining Phase II and now completed Phase III (M00-211, for a combined total of 1220 patients; Refs. 97, 149) reached statistical significance (but not when analyzed separately). Atrasentan appeared to be well-tolerated, with common side effects of headache, peripheral edema, and rhinitis. However, based on a September 13, 2005 meeting, the FDA’s Oncology Drugs Advisory Committee has recommended that Xinlay warrants further study and that it is not ready for final approval of this selective ERA-A for the treatment of metastatic HRPC even though the “drug has activity and was very highly likely to benefit”. Concerns were also raised to more clearly characterize potential cardiovascular safety risk because in a Phase III trial (M00-211), there has been a fourfold increase in cardiovascular-related deaths in those patients treated with atrasentan compared to placebo; the numbers are n = 8 (or 2% out of 404 patients in the treated group) vs. n = 2 (or 0.5% out of 397 patients in the placebo group). A blinded Phase III study in nonmetastatic HRPC (M00-244 with 941 patients) is ongoing and a blinded Phase II study in hormone-naïve prostate cancer (M01-366 with 222 patients) has recently been completed (Table 5). Additional open label studies are completed (M97-739) or ongoing (M00-258, M01-304 and M03-655; see below and Table 7). All eight Phase II/III substudies will have recruited a total of 3449 patients with prostate cancer. The ongoing M03-655 atrasentan study is an ongoing Phase I trial in combination with Taxotere (docetaxel) and/or prednisone evaluating the pharmacokinetic and pharmacodynamic drug interactions between these classes of compounds. Thus, atrasentan (Xinlay) constitutes an oral, nonhormonal, nonchemotherapy anti-cancer agent that may represent a new therapeutic option either as monotherapy or in combination with traditional chemotherapy. Abbott is continuing to support studies in other types of cancers, especially in ovarian cancer, renal cancer and glioma (Table 4; see below).
Atrasentan has also been in Phase II trials for hypertension, but development for this indication was halted in 2001 (86).
A pre-“CONSCIOUS-1” Trial with Clazosentan.
The pre-“Clazosentan to Overcome Neurological Ischemia and Infarct Occurring after Subarachnoid Hemorrhage (CONSCIOUS)” study is a Phase II-a study that has just been completed investigating whether clazosentan prevents cerebral vasospasm in patients following aneurysmal subarachnoid hemorrhage (Ref. 97; Tables 2 and 5), and its safety and tolerability profile. Treatment reduced the incidence and severity of angiographically-evident cerebral vasospasm as well as the incidence of new infarctions.
RAPIDS-1 and 2 with Bosentan.
Randomized Placebo-Controlled Study on Prevention of Ischemic Digital Ulcers in Scleroderma (RAPIDS)-1 was a 16-week study in 122 patients with ischemic digital ulcers with scleroderma. Patients receiving bosentan had a 48% reduction in the mean number of new digital ulcers during the study period (16).
RAPIDS-2 was a long term (24-week) Phase III clinical trial in 180 patients with ischemic digital ulcers with scleroderma. This study in patients with scleroderma was related to previous trials (see BREATHE-1, REACH-1, RAPIDS-1, and BUILD-2). Preliminary analysis of the RAPIDS-2 indicates that the primary endpoint of reduction in the occurrence of new digital ulcers during the 6-month treatment period was statistically significant. This result confirms the positive findings of RAPIDS-1. RAPIDS-2 also evaluated the effect on time to healing of existing digital ulceration in this patient population. There was no difference in time to healing between patients receiving placebo and those receiving bosentan. The safety profile of bosentan in this study was comparable to that observed in previous clinical trials and postmarketing experience with bosentan.2
STRIDE-1 to STRIDE-6.
Early nonpivotal clinical studies with sitaxsentan included: (i) a double-blind, placebo-control, acute hemodynamic study in 48 CHF patients (103), (ii) a 31-patient trial in essential hypertension (104), and (iii) an open-label trial in 20 patients with PAH (including six children; Tables 2, 5 and 7; Refs. 105, 111).
The first large-scale randomized clinical trial (STRIDE-1) by Encysive was aimed at patients with pulmonary arterial hypertension (PAH; Tables 2, 5 and 7). Overall, six STRIDE studies have targeted primary and secondary PAH patients, with doses of sitaxsentan ranging from 50 to 300 mg (150). In addition, four long-term extension studies, using sitaxsentan in patients for up to 58 weeks, have been completed (150). A small 1-year follow-up study to STRIDE-1 showed persistent improvement (151). STRIDE-6 (an open-label pilot) was designed to evaluate sitaxsentan treatment for patients with PAH who were discontinuing bosentan treatment because of safety or efficacy failure. The company completed the submission of an NDA with the FDA in May 2005 for Thelin (sitaxsentan) 100 mg as once-daily oral treatment for patients with PAH. The NDA contains the largest database ever assembled in a regulatory filing for PAH, with approximately 900 PAH patients receiving Thelin in clinical evaluations. In August 2005, the European Agency for the Evaluation of Medicinal Products accepted for review the company’s Marketing Authorization Application for Thelin. It is unclear to date whether selectivity for the ETA-R confers superior effects against PAH compared to dual ERA. The safety profile (e.g., incidence of liver enzymes abnormalities) might prove to be better. Only a direct head-to-head comparison (dose-related efficacy with a surrogate marker, patient selection, and primary and secondary end points) would answer this issue.
YM598 by Astellas Pharma (Formerly Yamanouchi).
In a Phase II multicenter clinical trial, YM598, added to mitoxantrone and prednisone, failed to control pain in hormone-refractory prostate cancer and prostatic neoplasms. The first record was received in November 2002 and terminated in June 2005, with no significant signs of improvement in the pain associated with prostate cancer metastases in the bone (Tables 2 and 5; www.clinical-trials.gov).
ZD4054 by AstraZeneca.
In a randomized placebo-controlled trial in eight healthy subjects, a single oral dose of ZD4054 reduced forearm vasoconstriction in response to brachial artery infusion of ET-1, thus providing clinical relevance of ETA blockade. ETB blockade was also assessed in an ascending, single-dose, placebo-controlled trial in 28 volunteers. For all doses of ZD4054, mean plasma ET-1 concentrations measured at 4 and 24 hrs were within the placebo reference range (indicating no significant ETB receptor blockade). There was no evidence of dose-related changes (Tables 2 and 5; Ref. 108). ZD4054 was then developed for prostate cancer with bone metastases. A Phase II-a trial has also been completed (see Tables 2 and 5).
Ongoing Clinical Trials or Trials Currently Recruiting Patients with Dual ERAs (Tables 2 and 6).
ASSET-1 and -2 with Bosentan.
The use of ERAs is being investigated in both primary and secondary forms of PAH (see Tables 4–7). Actelion is assessing the efficacy and safety of bosentan in patients with PAH related to sickle cell disease (SCD), a hemoglobinopathy, in the absence of left ventricular dysfunction. Assessment in Patients with sickle cell disease of the Efficacy and Safety of Bosentan Therapy on Pulmonary Hypertension (ASSET) will be a Phase III trial with 80, then 160 patients (Table 6). Sickle cell disease is characterized by chronic hemolysis, frequent infections, and recurrent vaso-occlusions of microcirculation, which cause painful crises and result in chronic vascular and organ damage and failure. Several observations indicate that ET-1 may be a key element of the pathogenesis of SCD (152, 153).
BENEFIT with Bosentan.
Bosentan Effects in Inoperable Forms of Chronic Thromboembolic Pulmonary Hypertension (BENEFIT) is a randomized, placebo-controlled, Phase III trial aimed at determining the efficacy and safety of bosentan in patients with chronic thromboembolic pulmonary hypertension (CTEPH). Various forms of pulmonary embolism are associated with PAH for which the role of the ET system has recently been reviewed (154).
Already, two open-label multicenter pilot trials have been conducted in 19 and 16 inoperable cases of CTEPH (155, 156). The first study found that following 3 months of treatment with bosentan, PVR decreased, whereas functional class and peak maximum rate of oxygen consumption remained unchanged. However, the 6MWD increased. Treatment was well tolerated by all patients. In the second study, patients were treated over 6 months. NYHA functional class was improved by one class in 11 patients. Mean 6MWDs increased. Regardless of uncontrolled design and small sample size, bosentan may offer a therapeutic option for patients with inoperable CTEPH.
BREATH-2 with Bosentan and Flolan.
This Phase II randomized, double-blind, placebo controlled trial will be conducted by CoTherix in 60 patients (Table 6), with a treatment arm combining bosentan with existing prostanoid therapy (iv epoprostenol (Flolan)). The combination of ERAs (at a lower dose) with established therapeutic agents has always been on the agenda toward increasing efficacy and reducing the extent of side effects. Other future options to be considered and tested include investigating the effects of combined treatment of bosentan with other prostacyclin preparations such as Treprostinil (sc) or Beraprost (inhaled Iloprost; see below).
Toward that end, a prospective, nonrandomized, open-label study in a university hospital setting has recently been reported. The addition of bosentan led to an increased exercise capacity (6MWD) and right ventricular function (Tei index) in 16 patients with PAH already receiving either Beraprost (inhaled Iloprost) or Iloprost iv at 6 months after initiation of combination therapy, and every 3 months thereafter (157). Another limited study revealed that bosentan was effective at replacing inhaled Iloprost, as reflected by early and sustained positive hemodynamic changes, precluding the return to previous therapy (158). This case report (n =2) was the first to document a switch of that sort.
BREATH-3 with Bosentan.
The safety and efficacy of bosentan therapy in children suffering from severe PAH has been demonstrated in two retrospective observational studies (159–161). The group of Maiya et al. (159) reported that in 40 children (mean age of 9.3 years, range 0.6–16 years) with various forms of PAH (class III and IV), bosentan was well tolerated and helped stabilize the children. However, parenteral epoprostenol was also needed in 60% of cases. In another retrospective study recently published (160), 86 children with PAH (idiopathic, associated with congenital heart or connective tissue disease) started bosentan with or without concomitant intravenous epoprostenol or subcutaneous Treprostinil therapy. Bosentan improved WHO functional class in 46% of cases and also decreased mean PAP and PVR. In addition, overall it was safe and efficacious for the treatment of PAH in children after a median exposure of 14 months.
Actelion is now planning a large Phase III trial of bosentan treatment in children with PAH.
BREATH-5 with Bosentan.
A Phase III clinical trial has been conducted with bosentan looking at PAH in patients associated with Eisenmenger’s syndrome (e.g., with congenital heart disease [CHD]), a target identified in 1993 by Cacoub et al. (Tables 2 and 6; Ref. 162). Initial experience with bosentan in such patients (n = 9, started on 62.5 mg/day for 4 weeks, than 125 mg bid; Ref. 163), revealed that six of nine patients (67%) had an improvement in NYHA classification and that oxygen saturation levels increased. In another open-label pilot study in 10 patients, similar results were obtained (164). The first open-label, prospective, multicenter trial (Phase II-a; n =33 patients/n = 23 with the syndrome) revealed that, after 2.1 ± 0.5 years of treatment, bosentan increased 6MWD and improved NYA class, in association with slight trends in improvement of transcutaneous oxygen saturation and maximum oxygen uptake, as well as decreased right ventricular systolic pressure (165).
BUILD-1 and -2.
Bosentan Use in Interstitial Lung Disease (BUILD) is a Phase III clinical trial that will investigate the safety and efficacy of bosentan in patients suffering from idiopathic pulmonary fibrosis (IPF), a chronic fibrosing lung disease limited to the lungs, with an estimated prevalence of 35,000–55,000 annual cases in the United States. Conventional therapy (corticosteroids, azathioprine, cyclophosphamide) provides only marginal benefit. Emerging strategies to treat patients with IPF include agents that inhibit epithelial injury or enhance repair, anticytokine approaches, agents that inhibit fibroblast proliferation or induce fibroblast apoptosis, and other novel approaches such as ERAs. The BUILD program with bosentan in patients suffering from either IPF (BUILD-1) or pulmonary fibrosis related to systemic sclerosis (BUILD-2), showed no effect on the primary endpoint of exercise improvement as measured by the 6-min-walk test (MWT). In the IPF study, BUILD-1, although not statistically significant, positive trends were observed for predefined secondary endpoints, such as the combined incidence of death or treatment failure at 12 months (36.1% in the placebo group vs. 22.5% in the bosentan group; P = 0.076; 95% confidence limits (CL) 0.37, 1.05), representing a relative risk reduction of 38%. Treatment failure (per protocol) was defined as worsening in pulmonary function tests (PFTs) or acute decompensation of IPF. Actelion intends to pursue a mortality-morbidity Phase III study.
COMBI.
Combination of Bosentan and Iloprost (COMBI) is a Phase IV trial, reminiscent of the BREATH-2 study, is to be conducted at the Hannover Medical School with 72 patients. It will assess the combined efficacy of bosentan with existing Iloprost in patients with iPAH (Table 6).
COMPASS-1 and −2 with Bosentan.
Combination of Pulmonary Arterial Hypertension Sildenafil Study (COMPASS) is another study aiming at assessing the combined efficacy of bosentan with another drug likely to be effective in patients with iPAH, sildenafil (Table 6). The first study will look at the hemodynamic effects in 40 patients, and the second study will look at morbidity and mortality in 600 patients (Table 6).
EARLY with Bosentan.
Endothelin Antagonist Trial in Mildly Symptomatic PAH Patients (EARLY) is a randomized, double-blind, placebo-controlled, multicenter study that will assess the efficacy, safety, and tolerability of bosentan in patients with mildly symptomatic pulmonary arterial hypertension. The Phase III trial constitutes a long-term (24 months) study in 170 patients presenting with less severe cases of PAH (functional NYHA class II).
FUTURE-1 and -2 with Bosentan.
The Pediatric Formulation of Bosentan in Pulmonary Arterial Hypertension (FUTURE) studies are 12-week trials that will assess the efficacy and safety of new formulations of bosentan in children.
Other Studies with Bosentan.
Thus far, the field of oncology, with regard to the applications of ET-related blockers, has belonged to Abbott and AstraZeneca, with atrasentan and ZD4054, respectively. Bosentan, as a dual ERA, has never been tested in such conditions. Because the incidence and already high mortality rates of malignant melanoma (a neoplasm) have been steadily increasing in recent decades (overall survival for patients with metastatic melanoma ranges from 4.7 to 11 months), Actelion has chosen to study this condition. This open-label pilot trial is aimed at determining the efficacy and safety of bosentan in patients with metastatic (malignant) melanoma, in combination with dacarbazine (DITC; 5-(3,3-dimethyl-l-triazeno)-imidazole-4-carboxamide, an antitumor agent) (Table 6).
Ongoing Clinical Trials or Trials Currently Recruiting Patients to Test Selective ERA-A (See Tables 2 and 7).
ARIES-1 by Myogen.
In November of 2005 Myogen announced the achievement of target enrollment of 186 patients in the Ambrisentan Randomized Multicenter Efficacy Study (ARIES-1), the last of the company’s two pivotal Phase 3 trials of ambrisentan in patients with PAH. A number of patients are completing the treatment phase of the study and will be allowed to be randomized into the extension trial (ARIES-E) in accordance with the trial protocol. The ARIES-1 and ARIES-2 studies were two Phase III, randomized, double-blind, placebo-controlled, multicenter, efficacy, and safety studies in subjects with PAH (Table 7). These studies were identical except for the doses of ambrisentan and the geographic locations of investigative sites. The doses selected for the ARIES-1 study were 5 and 10 mg per day, while ARIES-2 evaluates doses of 2.5 and 5 mg per day. Each study was 12 weeks in duration and enrolled approximately 186 subjects (62 subjects/arm). The primary efficacy endpoint was the change from baseline in the 6MWD evaluated after 12 weeks of therapy compared to placebo. Secondary endpoints included Borg dyspnea index, WHO functional class, the SF-36™ health survey, and time to clinical worsening. The company expects to report top line results of the trial in the second quarter of 2006. ARIES-2 now completed during the third quarter of 2005 (see previous section) showed significant improvement in 6MWD and in time to clinical worsening with no observed liver function abnormalities (Table 5).
ARIES-E by Myogen.
AMB-320/321-E is a Phase III, multicenter study examining the long-term safety and efficacy of ambrisentan in PAH subjects who have participated in ARIES-1 or ARIES-2. Eligible subjects will remain blinded and will continue to receive their last ambrisentan dose assignment from the previous study. However, subjects who received placebo during AMB-320 or AMB-321 will be randomized to active treatment (2.5 mg, 5 mg, or 10 mg). After week 24, the randomized treatment assignment will remain blinded, but investigators will be allowed to adjust study drug dose (available doses are 1, 2.5, 5, and 10 mg). After the clinical databases have been locked for both AMB-320 and AMB-321, subjects who have completed the week 24 visit will be unblinded and the ambrisentan dose may be further adjusted as clinically indicated. The primary endpoint of this study is the incidence and severity of adverse events associated with long-term exposure to ambrisentan in subjects with PAH. This study will also continue to evaluate the efficacy endpoints from the previous studies, as well as evaluate long-term failure-free treatment status and long-term survival.
AMB-222 by Myogen.
The incidence of liver function test abnormalities is being evaluated in a Phase II, open-label, multicenter study of ambrisentan in approximately 36 subjects with PAH who had previously discontinued bosentan or sitaxsentan (or both) therapy because of serum aminotransferase abnormalities. The primary endpoint of this study is the incidence of confirmed serum aminotransferase concentrations (AST or ALT) > 3 × ULN (upper limit of normal) during 12 weeks of ambrisentan therapy that are related to ambrisentan and resulted in discontinuation of the drug. The primary endpoint treatment period will be completed for all subjects by January 2006.
AMB-220-E by Myogen.
Fifty-four subjects who completed AMB-220 continued treatment in a Phase II, long-term, open-label study (AMB-220-E). Treatment effects observed during AMB-220 have been sustained for at least 48 weeks of ambrisentan therapy, including a greater than 50-m improvement in 6MWD, a significant decrease in dyspnea, and approximately two-thirds of subjects’ maintaining WHO Class I or II symptoms. In addition, 1-year and 2-year survival for PAH subjects was 92% and 89%, respectively, compared to 73% and 62% as predicted by an NIH Registry formula (93, 94).
GRH01 with TBC 3711 by Encysive.
This is a dose-ranging Phase II-a clinical trial initiated in 2005 in patients with resistant hypertension (Table 7). Darusentan (another ERA-A), and a Phase II-b trial named DAR-201 was previously conducted in patients with resistant hypertension (see previous section).
M00-244, M00-258, M01-304 and M03-655 with Atrasentan by Abbott.
It is established that the ET system is implicated in the regulation of cell proliferation, tumorigenic activities, cell death (apoptosis) and the formation of new blood vessels (angiogenesis) in renal, ovarian, brain, bone (abnormal osteogenesis) and non–small cell lung cancers (see Ref. 88 for a review).
In May 2002, Phase II trials for renal, ovarian, lung, colorectal, breast and brain cancers were commenced. Phase III trials (M00-244 and M00-258) were initiated in men with nonmetastatic prostate cancer (Table 7). A Phase II–III was also launched (M01-304) in men with rising prostate-specific antigen (PSA) following prostate cancer surgery (Table 7). Finally, a Phase I trial was initiated (M03-655) to assess drug interactions and pharmacokinetics in patients with prostate cancer who are showing progression (in combination with Taxtotere [docetaxel] and prednisone; Table 7).
Avosentan by Speedel.
Having completed Phase I and II-b studies (see above; Table 4; Ref. 44), Speedel is now running a pivotal morbidity and mortality Phase III trial in patients with diabetic nephropathy (see Table 7), a major cause of end stage renal disease accounting for about 40% of all new cases in the United States. Avosentan may have a positive effect on lengthening the time to doubling of serum creatinine and to end stage renal disease in these patients.
CONSCIOUS-1 with Clazosentan.
This Phase II-b trial involving 400 patients follows up the observations reported by Vajkoczy et al. (98) for Phase II-a. The profile of this Actelion study is reported in Table 7.
ZD4054 by AstraZeneca.
The recruitment of 250 patients worldwide started in September 2005 and was to be completed by November 2005 for a Phase II-b trial wherein patients with prostate cancer are to be divided in three groups (placebo, low dose of ZD4054, and high dose of ZD4054) for 2 years (www.CancerHelp.org.uk).
Safety and Pharmacotoxicology of ERAs
General Symptoms (Headache, Syncope, Flushing, Nausea, Rhinitis, Hypotension).
ERAs have symptomatic adverse effects that are typical for vasodilating agents. In general, the most frequent of these effects are headache and rhinitis. Other side effects include nausea with infrequent vomiting, postural hypotension, and flushing. These symptoms typically do not require discontinuation of the medication or dosage adjustment (the symptoms are not usually dose-dependent). Dual ERAs with these side effects include bosentan (at the dose of 125 mg bid used in most trials), avosentan (21, 90), darusentan (10–300 mg/day; Refs. 99, 100), and intravenous tezosentan (adverse effects were more frequent at higher doses; Refs. 54, 58, 59). ERA-A have similar adverse actions that do not appear to occur with any obvious difference in frequency than dual ERAs. Ambrisentan (93) and atrasentan most commonly causes rhinitis and headache that is not dose-related (except at very high doses of atrasentan; Refs. 95, 166). Initial studies with sitaxsentan at higher doses (4–6 mg/kg body weight/day) caused a relatively high incidence of rhinitis, headache, nausea and flushing (50–75%; Ref. 105). The drug was better tolerated with respect to these symptoms in subsequent studies using lower doses (100 or 300 mg/day; Ref. 107). Clazosentan given intravenously for up to 6–8 hr did not cause an increase in any of these side effects.
Edema and Congestive Heart Failure.
Both ERA-As and dual ERAs cause fluid retention. Bosentan therapy was associated with early worsening of CHF (within the first 4–8 weeks) in the ENABLE and REACH-1 trials (51, 52) and this was felt to be a consequence of fluid retention. In the EARTH and HEAT-CHF trials, darusentan tended to worsen CHF when given at higher doses (99, 100). Most importantly, only in the short-term HEAT-CHF trials, darusentan administration was associated with an increased death rate (placebo, no deaths; 30 mg, no deaths; 100 mg; 2 deaths [5.1%] in 39 patients; 300 mg, 2 deaths [4.1%] in 29 patients; Ref. 100). Intravenous tezosentan, at doses greater than 1 mg/hr, reduced urine output in patients with acute CHF, thereby limiting clinical efficacy (74, 167). Interestingly, selective ERA-As also cause fluid retention. Ambrisentan (1–10 mg/day) is associated with edema (25% incidence; Ref. 93). Sitaxsentan at higher doses significantly causes edema (105), whereas lower doses (100 or 300 mg daily) have less tendency to cause edema (107). In initial studies in patients with metastatic prostate cancer, atrasentan caused a dose-dependent increase in edema (168), whereas similar edema side effects were seen in patients with various advanced malignancies (166). In this latter study, two patients on a very high dose of atrasentan (75 mg/day) developed severe hyponatremia. Patients given lower doses of atrasentan (2.5 or 10 mg daily) still had a 33% incidence of peripheral edema that was associated with a 1 kg gain in body weight (95).
Anemia.
Several ERA-As and dual ERAs cause a mild anemia within the first few weeks of therapy that stabilizes after about 4–8 weeks despite continued therapy. Although red blood cell mass has not been studied, it has generally been assumed that the anemia is because of hemodilution because it is typically associated with a modest weight gain and peripheral edema. Agents that have been reported to cause edema include bosentan (hemoglobin [Hgb] fall of 0.9 g/dl; Refs. 45–47, 169), ambrisentan (Hgb decrease of 0.8 mg/dl; Ref. 93), sitaxsentan (Hgb decreased 1.0 mg/dl on a dose of 100 mg of sitaxsenten/day and 1.6 mg/dl on 300 mg/day; Ref. 107), and atrasentan (Ref. 95). Avosentan given for only 7 days did not change Hgb, although this was likely not long enough to see an effect (90).
Abnormal Liver Function Tests.
Most ERAs are associated with elevations in hepatic transaminases. Initial studies with bosentan in PAH revealed a higher incidence of aminotransferase abnormalities in patients using the highest dose (45–47). Because there was no difference in efficacy, the lower dose (125 mg bid) was used for subsequent studies. In a 15-month follow-up, 15% (3/21) of patients had increased transaminases that did not lead to drug discontinuation (169). In contrast, in the RAPIDS trial, 5 of 79 patients treated with bosentan for systemic sclerosis developed abnormal liver function tests that required drug discontinuation; liver function subsequently normalized (170). Other ERAs cause transient increases in liver transaminases, including avosentan (seen with higher doses during a 7-day study; Ref. 90) and darusentan (2 of 39 patients on 100 mg/day; Ref. 100). Ambrisentan caused modest increases in hepatic transaminases over a 12-week period (2 of 64 patients; Ref. 93). In early studies using very high doses (4–6 mg/kg body weight/day), sitaxsentan caused a 35% incidence of elevated hepatic transaminases (20 patients total), two of whom developed severe hepatitis that was fatal in one case (105). The number of patients who showed increased liver aminotransferases using 100 and 300 mg sitaxsentan once daily during STRIDE-1 (12 weeks) and STRIDE-1X (mean follow-up of 26 weeks), were: STRIDE-1, placebo 2/59, 100 mg 0/56 and 300 mg 6/63; and STRIDE-1X, 100 mg 4/77 and 300 mg 19/91 (107). In STRIDE-2, sitaxsentan 100 mg once daily was associated with a 3% increase in liver function abnormality in the 18-week study, compared to 11% for bosentan and 6% for placebo. Furthermore, the Kaplan-Meier estimate of time to abnormal liver function test (LFT) elevation at 1 year of exposure was 4.0% for sitaxsentan 100 mg and 18.7% for bosentan (P = 0.0086; Ref. 171). Thus, these data suggest that the incidence of liver function abnormalities may be significantly different between the various ERAs.
Drug Metabolism.
Bosentan, a mild inducer of CYP450 2C9 and 3A4, decreases simvastatin, cyclosporine, warfarin, and glibenclamide levels (172, 173). Ketoconazole and cyclosporine increase bosentan levels (173). Bosentan and glibenclamide in combination are contraindicated because of increased risk of aminotransferase elevations (174). Drug interactions with other ERAs have not been extensively reported.
ERAs have variable effects on INR (international normalized ratio related to prothrombin time). As stated above, bosentan decreased warfarin effectiveness, at least in one patient (175). In contrast, sitaxsentan increases the INR in humans because of its inhibition of CYP2C9 P450, the major hepatic metabolizer of warfarin (107). Ambrisentan, another ERA-A, does not affect INR (93).
Testicular Function.
There is no information in the published literature on the effects of ERAs on testicular function. Despite this, in the product literature for bosentan, it is stated that many endothelin receptor antagonists induce marked atrophy of the seminiferous tubules of the testes, reduce sperm counts, and decrease male fertility in rats when administered for longer than 10 weeks. It is also stated that these effects appear to be irreversible. In contrast, Actelion states that bosentan did not affect testicular structure or function unless it was given at high doses for very prolonged periods (2 years in rats). There have been no studies to date on the effects of any ERAs on testicular function in man.
Teratogenic Activity and Contraceptives.
All ERAs are contraindicated in pregnancy. In animal studies, disruption of endothelin receptor A or B isoforms during embryogenesis causes severe developmental abnormalities associated with perinatal lethality (175, 176). There is little information on the impact of ERAs on the efficacy of oral contraceptives. The most widely studied agent, bosentan, may alter metabolism of hormonal contraceptives. Actelion, as well as other makers of ERAs, advises women of childbearing age to use at least two forms of birth control, one of which is nonhormonal.
Summary and Conclusions
As put forward by Lee and Rubin (178), the lack of absolute significant improvement in clinical parameters in studies involving ERAs does not equate to: FAILURE (i.e., the latest Phase III trial conducted with atrasentan in prostate cancer). Rather than causing an overt improvement, ERA treatment might result in stabilization or slowing of the rate of disease progression. Studies over longer periods, albeit more costly, may reveal such effects.
Bosentan, the most studied, both preclinically and clinically, has a good safety profile (as shown by the TRAX database, made up of over 5000 treated patients in Europe) with an incidence of abnormal LFT of 7.4%. In long-term follow-up studies (after 1, 2, and 3 years), bosentan treatment improved survival (estimates of 96%, 89%, and 86%, respectively (179) when compared to the NIH-PPH registry equation predictor (180). Pediatric experience over 2 years has also revealed a 91% survival rate in this sensitive population (181). In the context of polypharmacy related to cases of PAH, Actelion is also pioneering trials to compare bosentan with various prostacyclin analogues and Sildenafil.
By detailing all completed, stopped, and ongoing clinical trials involving ERAs, in even further detail than what we are presenting here, lessons can be learned in terms of dosing, efficacy, toxicity, and receptor selectivity. In this way the design of new trials can be improved.
Perspectives
Preclinical studies conducted in several experimental animal models of disease suggest that a number of therapeutic targets involving the dysregulation of the ET system remain to be studied in clinical trials. Such targets include other forms of PAH associated with connective tissue diseases such as lupus, rheumatoid arthritis, derma-topolymyositis, and Sjogren’s syndrome (94). ERA could also be developed as a novel pharmacotherapy for chronic obstructive pulmonary disease, which is associated with pulmonary artery intimal thickening and vessel narrowing; chronic obstructive pulmonary disease has excess ET receptor expression and elevated circulation plasma concentrations of ET-1. Additional targets include cancer (bone, lung, ovarian); renal diseases (hepatorenal syndrome [in which indication, however, tezosentan was evaluated and stopped in the second quarter of 2003] and chronic renal insufficiency); and cardiovascular-remodeling conditions (various other forms of hypertension, including nonidiopathic related forms of PAH, CHF, and acute myocardial infarction). They also include obesity-related Type 2 diabetes and oxidative stress, respiratory-fibrotic-hypoxic diseases (allergic asthma, and also sarcoidosis-related PAH, for which bosentan may constitute an effective treatment; Ref. 182), pain control, various diseases of the eye, and subarachnoid hemorrhage (as detailed in articles in these Proceedings of the Ninth International Conference on Endothelins). ERAs might also offer benefit in preventing complications of certain treatments, such as nephrotoxicity following the injection of radio contrast, rejection after graft transplantation, restenosis postangioplasty, and cardiorenopathy and edema following administration of blood substitutes.
For instance, bosentan could be used as a bridging therapy before (heart)-lung transplantation in patients with severe left ventricular dysfunction secondary to iPAH; such a treatment might improve ventricular function, making the patient more amenable to transplantation (to the point that the heart component of the whole approach is no longer necessary; Ref. 183). ERAs could be added to transplant solution for organ stabilization and preservation, and also to reduce inflammation, prevent post-transplant ischemia-reperfusion injury and some of cyclosporine-mediated (or other antirejection therapy) side effects, nephrovascular-related side effects, post-transplant complications, and transplant failure.
For many of these potential applications, the presence of existing therapeutic options means that the financial incentives for pharmaceutical companies to launch expensive and decisive clinical trials are currently lacking. Furthermore, benefit from ERAs must be shown in addition to treatment with established therapy. For example, there is a reluctance to develop ERAs as an antihypertensive treatment, despite many patients having high BP that is resistant to multidrug treatment, though two “non-big pharmaceutical” companies are doing so. By learning lessons from previous clinical trials, summarized in this review, we hope that the design of future studies of ERAs in these new potential therapeutic areas will be improved.
List of Endothelin Receptor Antagonists (ET-Ra = ERA) that Underwent Extensive Preclinical Development Leading to Clinical Studies and/or Further Drug Development.
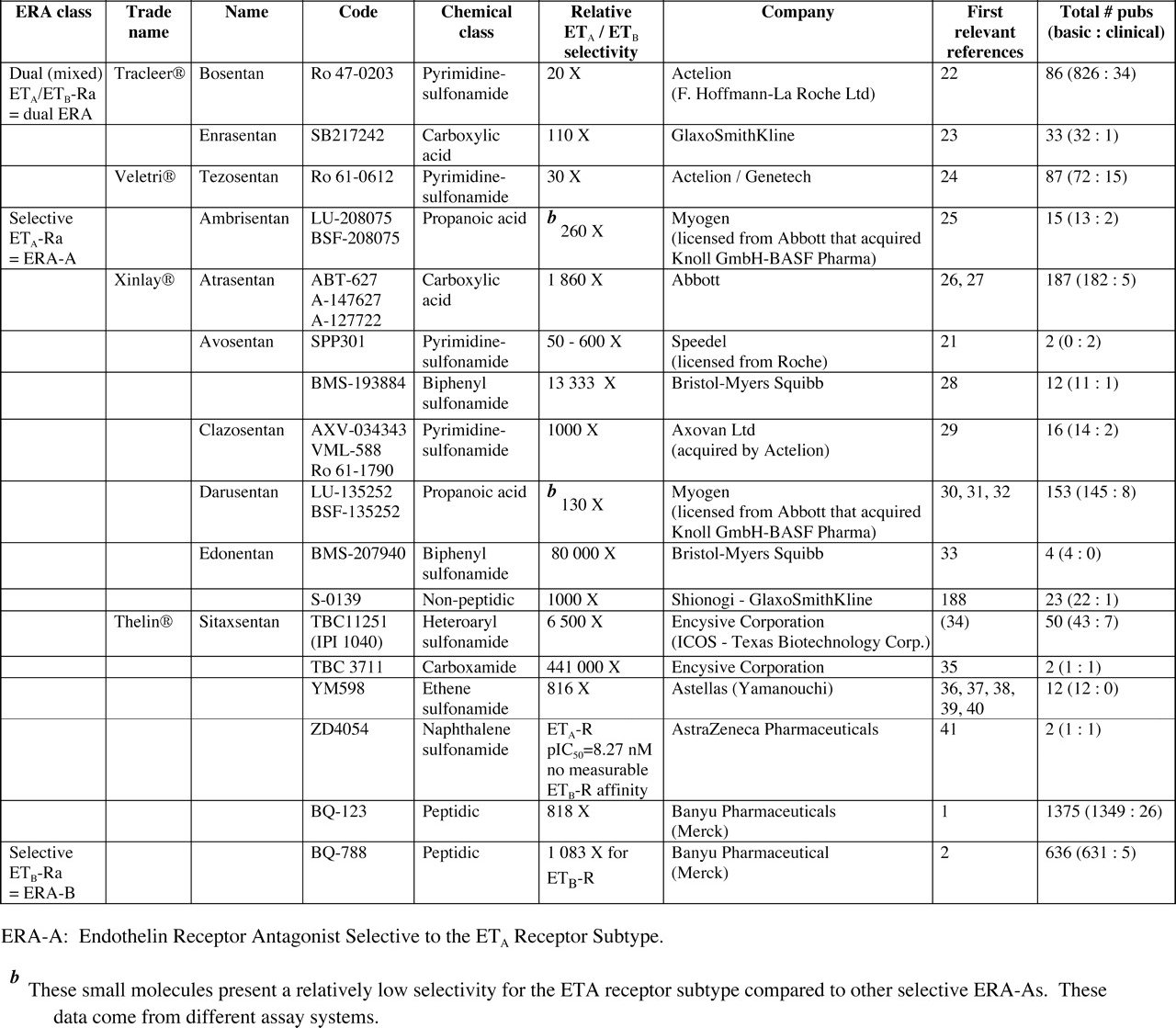
Summary of Clinical Trials Conducted Using an Endothelin Receptor Antagonist (ERA).

A Selection of Completed and Ongoing Clinical Academic Studies and Formal Trials Conducted with ET-1 and Endothelin Receptor Antagonists (ERAs) in Healthy Control Subjects and Patients.

Completed Clinical Trials Conducted with Dual Endothelin Receptor Antagonists (dual (mixed) ERAs).

Completed Clinical Trials Conducted with Selective Endothelin Receptor Antagonists to the ETA Subtype (ERA-A).
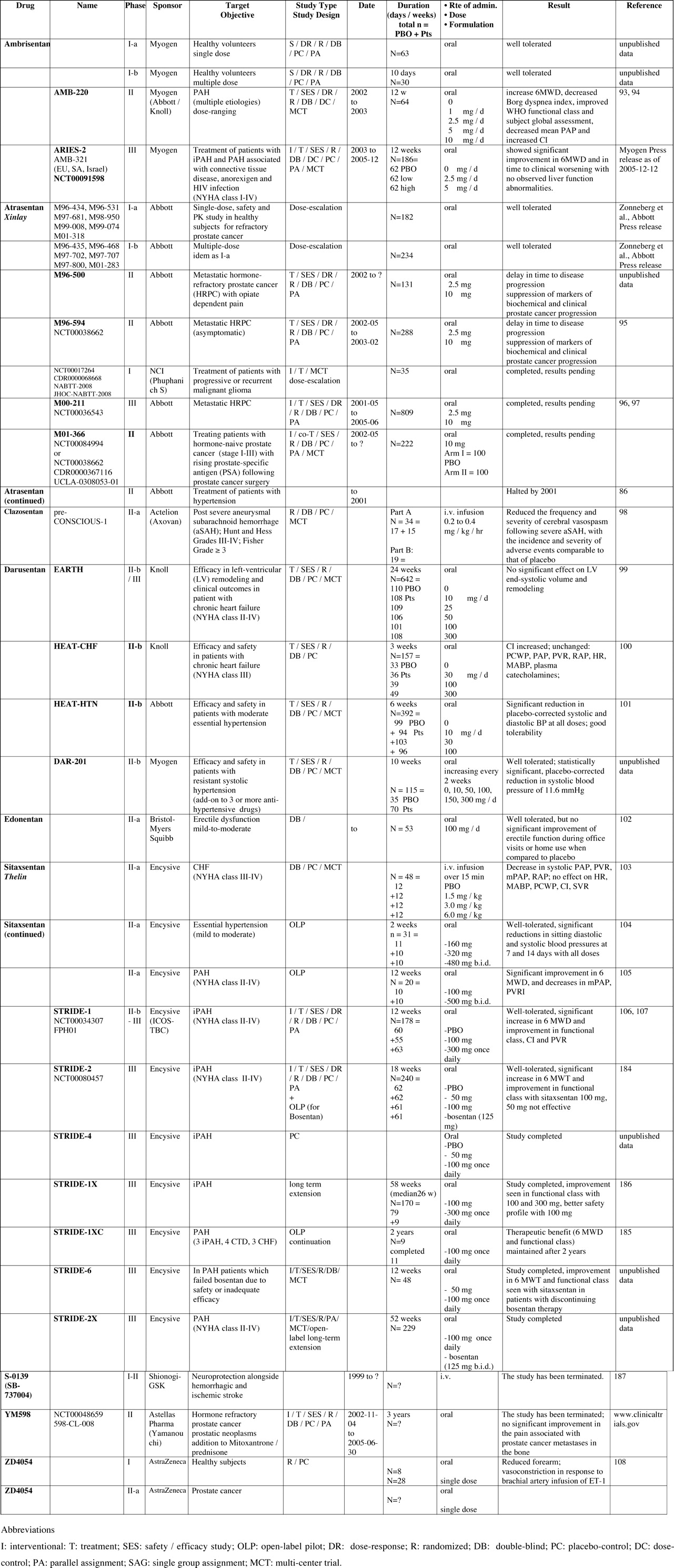
Ongoing and/or Planned Clinical Trial Studies Undergoing Recruitment of Dual Endothelin Receptor Antagonists (dual (mixed) ERAs).
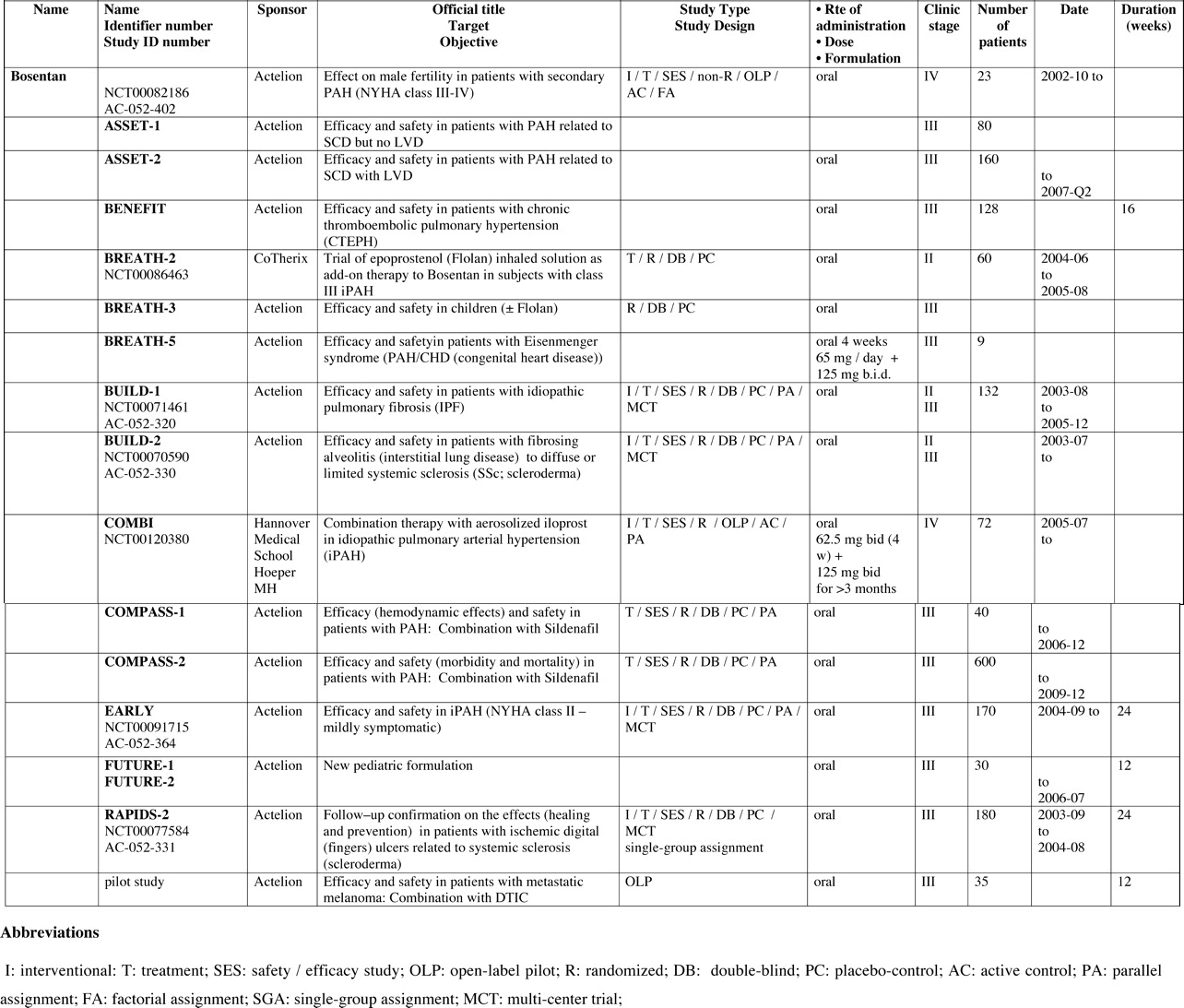
Ongoing and Planned Clinical Trial Studies Undergoing Recruitment with Selective Endothelin Receptor Antagonists (ERAs) of the A Subtype.
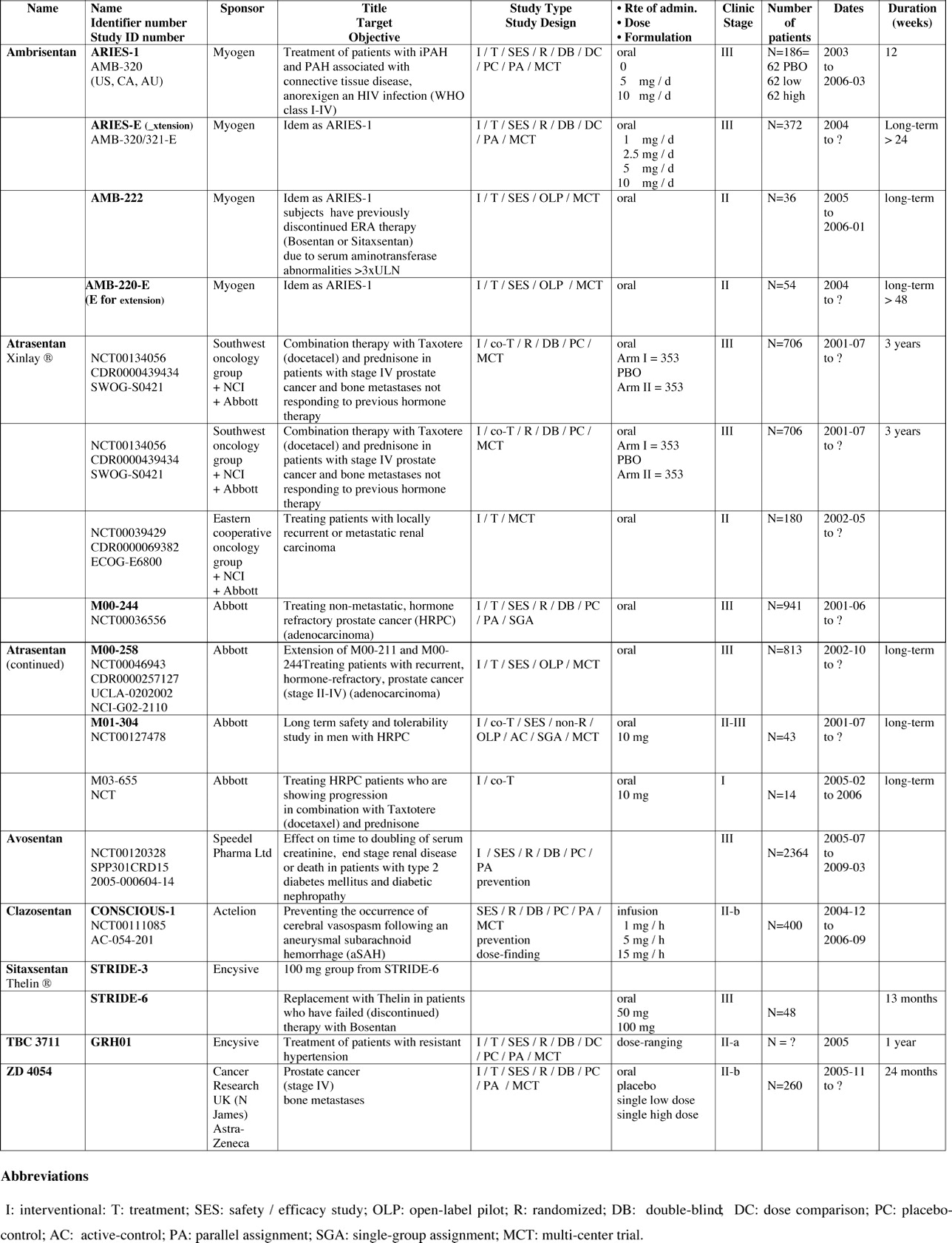

(a) Structures of the first generation of peptidic ERA.
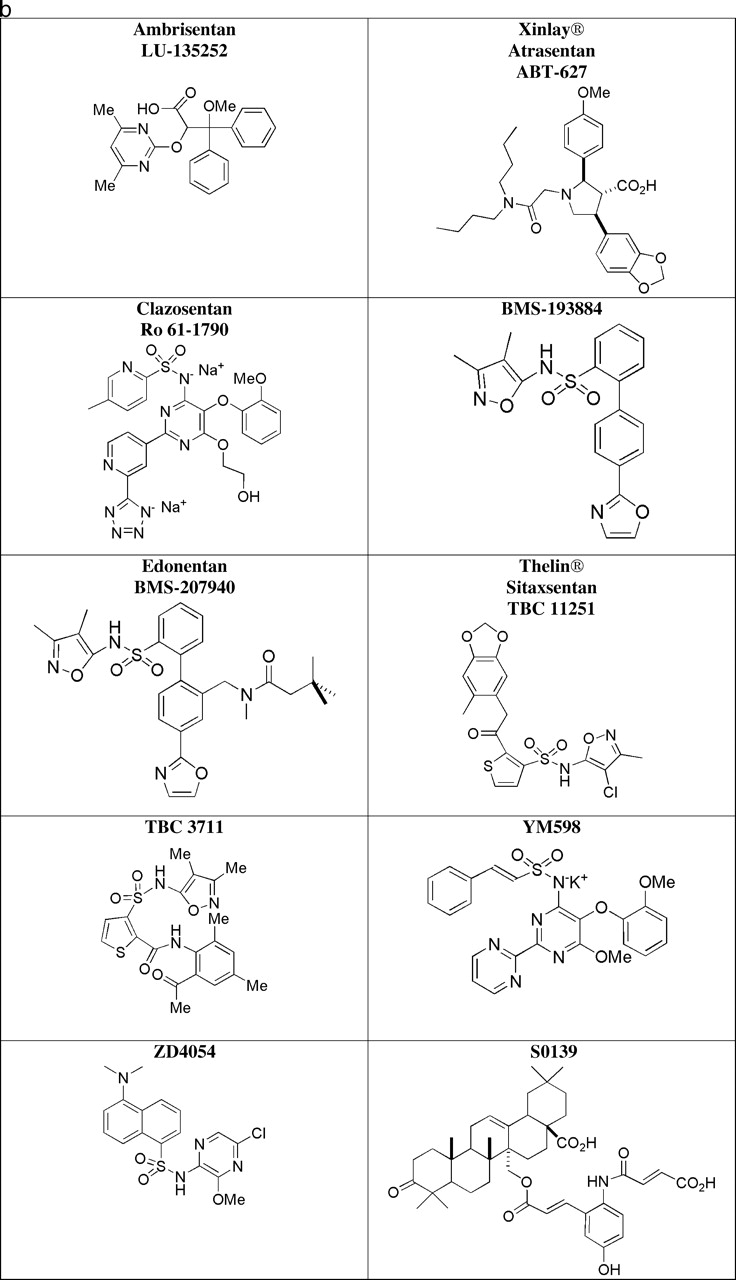
(b) Chemical structures of selective ERA-A.
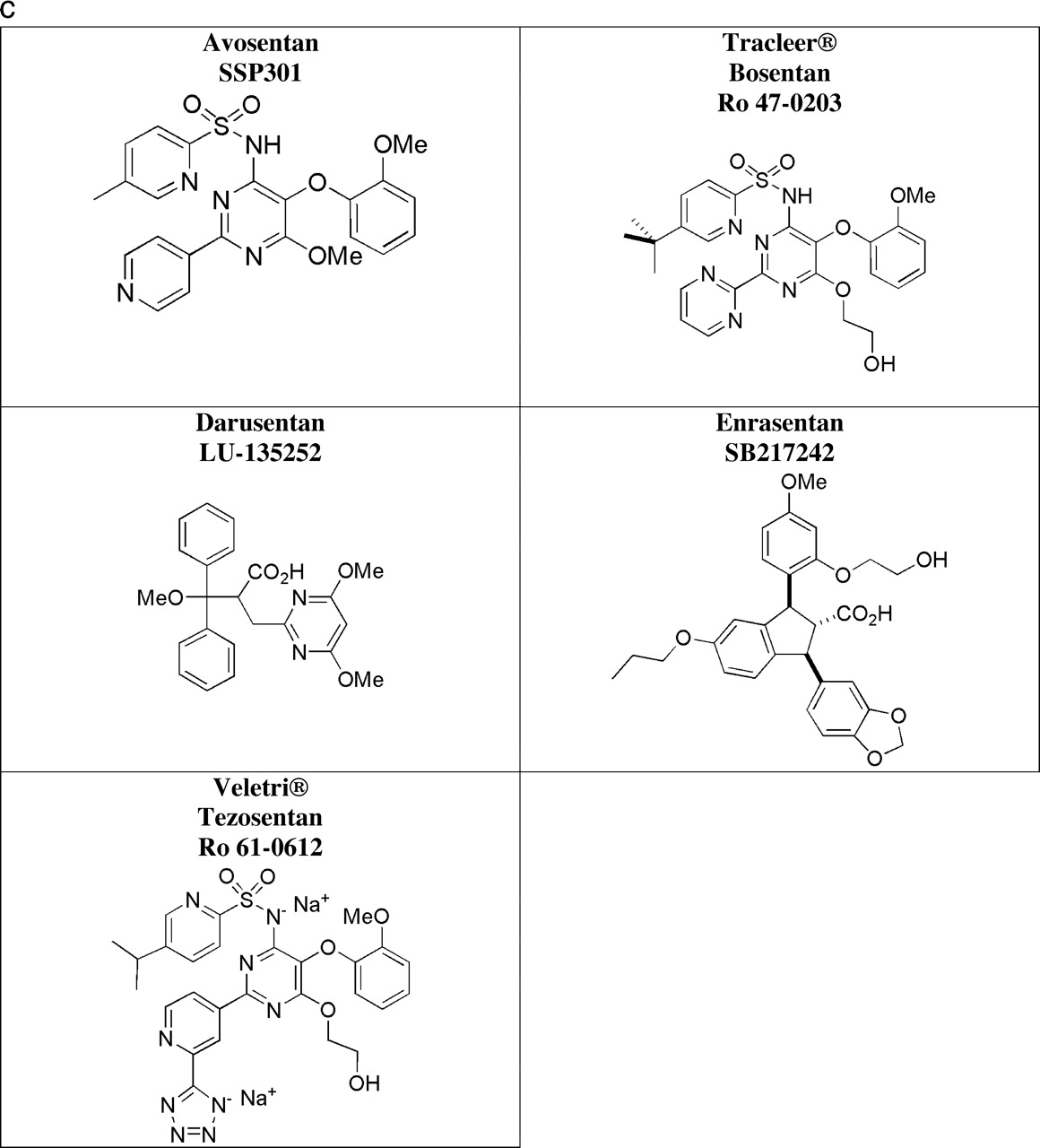
(c) Chemical structures of dual (mixed) ERA.
Footnotes
Acknowledgements
The authors would like to thank these individuals for their collaboration in reviewing and completing the tables: Martine Clozel and Marie-Claude Lefebvre at Actelion; Tom Brock and Richard Dixon at Encysive; Joel Nelson, Charles L. Van Sant, Joyce Steinberg, and Darryl J. Sleep at Abbott; Clive D. Morris at AstraZeneca; Jessica Mann at Speedel; and Richard J. Gorczynski and Robert L. Roden at Myogen.