
Abstract
Endothelin-converting enzyme (ECE)-1 cleaves big endothelins, as well as bradykinin and β-amyloid peptide. Several isoforms of ECE-1 (ECE-1a, 1b, 1c, and 1d) have been identified to date, they differ only in their amino terminus and share the catalytic domain located in the C-terminal end. In addition to full-length ECE-1 forms, we identified novel, alternatively spliced messenger RNAs (mRNAs) of ECE-1b, 1c, and 1d. These splice variants (SVs) lack exon 3′, which codes for the transmembrane (TM) region and is present in full-length forms. SV mRNAs were highly expressed in endothelial cells (EC) derived from macrovascular and microvascular beds. Analyses of ECE-1d and its SV forms in stably transfected human embryonic kidney (HEK)-293 cells revealed that both proteins were recognized by antibodies to C-terminal ECE-1, but an antibody to the N-terminal only bound ECE-1d. The novel protein, designated ECE-1sv, has an apparent molecular weight of 75 kDa. ECE-1sv lacks the TM sequence (or signal peptide) and, therefore, is expected to remain cytosolic. Presence of ECE-1sv in different cellular compartments than the full-length forms of the ECE-1 may suggest a distinct physiologic role for these proteins.
Introduction
Endothelin-converting enzyme (ECE)-1 is a Type II membrane protease that belongs to the neprilysin family of zinc metallopeptidases (1, 2). ECE-1 is abundantly expressed in the vascular endothelial cells (EC) of all tissues but is also found in nonvascular cells (3–6). This enzyme is characterized by a single transmembrane (TM) region, a short N-terminal cytosolic tail, and a large C-terminal extracellular domain that contains the enzymatic active site (7). ECE-1 is a glycosylated protein with 10 putative N-linked glycosylation sites (8). The best-characterized substrates are the endothelin (ET) family, consisting of three isopeptides, termed ET-1, ET-2, and ET-3, which are derived from distinct genes (9, 10). ET-1, the most abundant of the three, is a pleiotropic peptide; although best known for its vasoconstricting activity, it has diverse biologic functions (11–13). ETs are synthesized from an approximately 200–amino acid precursor, prepro-ET, after removal of their signal peptide, ETs are processed by dibasic pair-specific enzymatic activity to form the respective inactive big ETs (38–41 residues long; Refs. 1, 4). ECE-1 then specifically hydrolyzes the Trp21-Val/Ile22 bonds of big ETs to produce biologically active ETs (1, 14). Nevertheless, ECE-1 hydrolyzes several substrates other than big ET-1, suggesting that this enzyme may be involved in diverse biologic processes and pathologic conditions (15–17). Therefore, ECE-1 inhibitors are considered a useful therapeutic modality for a variety of disease states. Four isoforms of ECE-1 (1a, 1b, 1c, and 1d) have been identified, to date, in human, rat, and bovine (8, 18–21). Although the four proteins are encoded by one gene, each is expressed from a distinct promoter that regulates the expression of the four unique amino termini (8, 18–20). Although the ectodomain containing the active site is identical in each of the isoforms, the amino-terminal sequences seem to be responsible for differences in subcellular localization (18, 22–24). In this paper, we report the initial characterization of a novel splice variant (SV) of ECE-1 that lacks the TM domain.
Materials and Methods
Materials.
Dulbecco’s modified Eagle’s medium (DMEM) low glucose, DMEM with Ham’s F12 1:1 (v/v) nutrient mixture, SuperScriptII RNase H–reverse transcriptase, calf serum, and Ultra pure electrophoresis agarose gel were obtained from Gibco BRL Life Technologies (Gaithersburg, MD). Vitrogen and Type I collagen were obtained from Cohesion Technologies (Palo Alto, CA). Penicillin, streptomycin, and fetal calf serum (FCS) were obtained from Biological Industries (Beit Haemek, Israel). TRI reagent was obtained from MRC (Cincinnati, OH). Deoxynucleotide triphosphates, random hexamer oligodeoxynucleotides, and Taq DNA polymerase were obtained from Fermentas (Vilnius, Lithuania). Protease inhibitor cocktail for mammalian cell extracts, oligonucleotide primers, and horseradish peroxidase (HRP)–conjugated goat anti-rabbit IgG were obtained from Sigma (St. Louis, MO). Protein quantification kits were obtained from Bio-Rad Laboratories (Hercules, CA). Hifidelity Taq polymerase was obtained from Takara (Otsu, Shiga, Japan). Restriction enzymes were obtained from Fermentas (Hanover, MD). N-glycosidase F (PNGase F) was obtained from New England Biolabs (Beverly, MA). FuGENE 6 transfection reagent was obtained from Roche (Indianapolis, IN). pGEM-T vector, pcDNA6/V5-HiS version C, and Blasticidin were obtained from Invitrogen (Carlsbad, CA).
Cell Cultures.
Microvascular EC derived from the bovine corpus luteum (25–27), termed luteal EC (LEC) were grown in complete DMEM Ham’s F12 containing 10% FCS and 2 mM glutamine on plates precoated with 2% Vitrogen. Human umbilical vein EC (HUVEC) and the SV40-transformed luteinized human granulosa cell line (SVOG) were kindly provided by I. Vlodavsky of the Hadassah-Hebrew University Hospital (Jerusalem, Israel) and N. Auersperg (University of British Columbia, British Columbia, Canada), respectively. Human embryonic kidney (HEK)-293 cells were cultured in complete DMEM Ham’s F12 medium containing 10% FCS and 2 mM glutamine. SVOG and LEC cells were from passages 5–12, HUVEC were from passages 2–4. All cells were grown to 70%–80% confluence.
Production of bECE-1 Constructs.
The complementary DNA (cDNA) sequences of full-length bovine ECE-1d and ECE-1d SV were amplified with the 1d and ECE-1 ends as primers (Table 1). The amplification products were separated on agarose gels and the corresponding single bands were extracted and cloned onto pGEM-TEasy vector. Inserts were subsequently subcloned into pcDNA vectors (pcDNA6/V5) and sequenced. A shorter SVcut construct lacking the first 169 base pairs (bp) of SV was generated by digesting SV with Eco91I (BstEII). HEK-293 cells were transfected using the FuGENE 6 transfection reagent. Stably transfected cells lines (containing bECE-1d, SV, and SVcut) were established using 1 μg/ml Blasticidin as a selective antibiotic.
Western Blot Analysis.
The procedure for total cell extracts was carried out as we have previously described (28, 29). Briefly, cells were homogenized in lysis buffer (25 mM Tris HCl; 100 mM NaCl; 0.5% deoxycholate; 0.5% Nonidet P-40 (NP-40); 5 mM EDTA, pH 7.5; and 10% protease inhibitor cocktail). Cell extracts were sonicated on ice for 10 secs at low speed. Protein concentration was determined using Bio-Rad DC reagents. All steps were performed on ice and samples were kept at −80°C until use. Proteins were electrically transferred to nitrocellulose membranes. After 2 hrs of blocking in Tris-buffered saline–Tween 20 + 5% low-fat milk, membranes were incubated with the appropriate ECE-1 antibody. Antiserum to total ECE-1 (antibody to C-terminal) was raised against a synthetic peptide comprising the last 16 amino acids of ECE-1. The membranes were washed three times and incubated with HRP-conjugated goat anti-rabbit IgG for 1 hr at room temperature. A chemiluminescent signal was generated with SuperSignal and the membranes were exposed to x-ray film.
RNA Extraction and Reverse Transcriptase Polymerase Chain Reaction (PCR).
Total RNA was extracted from the cells using TRI reagent. One microgram of total RNA was reverse transcribed, and semiquantitative PCR was performed, as described previously (5, 30). The sequence of the primers used in the PCR reactions is shown in Table 1.
Deglycosylation by PNGase F.
Recombinant proteins (ECE-1d full and SV forms) underwent deglycosylation by PNGase F as follows: total protein extract from either HEK-293 cells expressing full-length ECE-1d or its SV form (20 μg) was incubated for 10 mins at 100°C in denaturing buffer containing NP-40, then incubated for 1 hr at 37°C without (mock treatment) or with 1500 U of PNGase F.
Results and Discussion
PCR amplification of cDNA derived from LEC with a 5′ primer specific for each ECE-1 isoform and a common reverse primer located approximately 500 bp downstream (Table 1) generated two products for isoforms 1b, 1c, and 1d (Fig. 1A). The upper bands were of the expected size and the lower ones seemed to be approximately 140 bp shorter. A similar pattern of ECE-1 isoform expression was obtained in bovine aortic endothelial cells (data not shown). The upper bands were the expected PCR products, based on the known sequences of bECE-1b, 1c, and 1d (21) and the lower bands were SVs lacking the same 142-bp sequence, corresponding to exon 3′, found in all known ECE-1 isoforms (Fig. 2; Ref. 20). No such SV (lacking exon 3′) was observed for the ECE-1a isoform, whose transcription begins further downstream, in exon 3. In SVOG (Fig. 1B) and HUVEC (Fig. 1C), the SV of ECE-1d is also clearly identified.
We next used two different sets of primers to detect the full-length forms of the enzyme versus the spliced variant forms. We designed primers that spanned a unique sequence that is present in SV (produced by the end of exon 2′ merged with exon 4; Fig. 2). Full-length ECE-1 forms were amplified with a primer that resides in the sequence of exon 3′, which is absent from SV forms (Fig. 2; Table 1). In both PCR reactions, the same reverse primer was used (common ECE-1; Table 1). With each set of primers, one band (Fig. 1D) was amplified, in agreement with the data shown in Figure 1A, the SV band was approximately 140 bp shorter.
The hitherto known isoforms of ECE-1 arise from the use of alternative promoters upstream of exon 3′ (18, 20). ECE-1sv is the first identified ECE-1 isoform that arises from an internal exon splicing. Alternative splicing is widespread in mammalian gene expression and is a major contributor to the functional complexity of mammalian genomes (31, 32). Whether the ratio of ECE-1 to its SV form also differs between different physiologic or pathologic conditions is, as yet, unknown; however, it is noteworthy that ECE-1sv was highly expressed in EC.
Because ECE-1d and its SV form were both abundant (Fig. 1) we cloned and stably expressed its full-length and SV forms. An additional cDNA, in which part of the 5′ end of SV was deleted (SVcut) was stably expressed in HEK-293 cells as well. An antibody that recognizes the common C-terminal end of the enzyme identified a protein product in all cells expressing ECE-1d, SV, and SVcut (Fig. 3). Cells transfected with full-length ECE-1d expressed, as expected, a protein of approximately 120 kDa, whereas SV- and SVcut-transfected cells both expressed a protein of approximately 75 kDa (Fig. 3). A different antiserum directed against the N-terminal part of ECE-1d readily recognized the full-length ECE-1d, but not the protein products of either SV or SVcut. These findings, therefore, suggest that SV forms share only the C-terminal end with ECE-1d. Because N-terminal antibodies did not recognize the translated products of SV and SVcut (Fig. 3), it seemed that these forms had a different start codon from that used in ECE-1d. This was, in fact, expected because splicing out the 142-bp segment would modify the reading frame. Because cDNAs of both SV and the shorter form, SVcut, were translated into proteins with the same apparent molecular weight, it was suggested that the start site of this protein was further downstream in a region common to all ECE-1 forms, so that the three messenger RNA (mRNA) species were translated into one protein, ECE-1sv. Because the catalytic domain of the enzyme is found in its far C-terminal end, the translation product is expected to retain its bioactivity.
Unlike ECE-1d, ECE-1sv, which lacks the TM (or signal peptide) portion, cannot be routed to the secretory pathway and is expected to remain nonglycosylated (Fig. 4). To test this assumption, the recombinant proteins were treated with PNGase F, which removes the N-linked oligosaccharides added in the Golgi complex. Indeed, the molecular mass of ECE-1sv was not affected by PNGase F treatment, in contrast to ECE-1d, in which deglycosylation reduced the molecular weight from 120 kDa to approximately 85 kDa (Fig. 4). It is noteworthy that deglycosylation of purified soluble ECE-1 did not significantly alter its enzyme activity (33), suggesting that cytosolic ECE-1sv may be biologically active. However, another study (34) showed that double mutation of two conserved N-glycosylation sites, Asn-632 and Asn-651, abolished the enzymatic activity; therefore, this issue awaits further investigation.
The catalytic site of full-length ECE-1 faces the extracellular milieu or the luminal side of vesicles, whereas the active site of ECE-1sv resides within the cells cytoplasm. What are the putative substrates of this unique isoform? It is tempting to speculate that cytosolic ECE-1sv may degrade small peptides, such as angiotensin I, bradykinin, neurotensin, and substance P, internalized via their receptors, or cytosolic ECE-1sv could also cleave ß-amyloid peptide. All of these peptides (15, 16) are very efficiently degraded by soluble ECE-1 engineered by truncating its TM domain. Breaking down these small peptides by ECE-1sv could terminate their signaling in a manner analogous to the action of insulin-degrading enzyme (IDE). IDE is a ubiquitously expressed neutral thiol metalloprotease that is present mainly in the cytosol and peroxisomes. The active site of IDE, HEXXH, is very similar to that of ECE-1, HELTH, which could explain the overlapping specificity of these enzymes, that is, cleavage of insulin and amyloid peptides.
Interestingly, the presence of a soluble intracellular form of ECE-1 in endothelial and vascular smooth-muscle cells was postulated in the past (21, 35); however, no molecular identification was provided before.
The mechanism responsible for ECE-1sv translation most likely involves re-initiation of translation. This was shown to occur with mRNAs that have small open reading frames (ORFs) near the 5′ end (36). These short ORFs are initiated at the respective start codons of isoforms ECE-1b, 1c, and 1d, and are terminated by a stop codon produced by splicing out exon 3′ (~50 codons). It is also noteworthy that the putative start codon that initiates the translation of ECE-1sv, ccATGg, has a strong Kozak’s motif (36).
The findings presented here, which demonstrate the plethora of different ECE-1 forms, full-length and spliced variants, that coexist within EC, offer a novel perspective on the physiologic activation of ET-1 and of other substrates of the ECE-1 family of proteins.
PCR Primer List
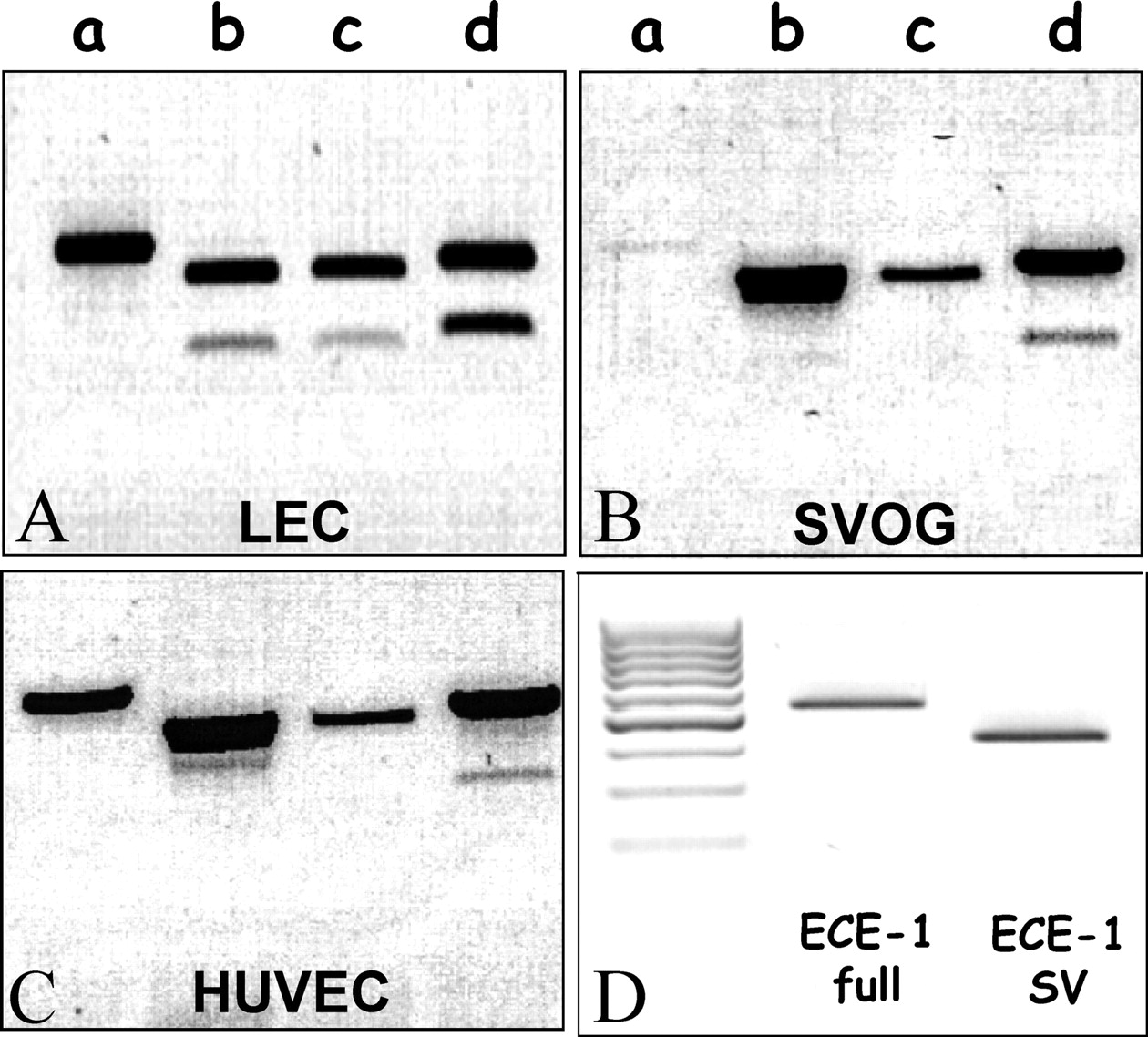
PCR amplification of full-length ECE-1 and alternatively spliced isoforms in SVOG (A), bovine LEC (B), and HUVEC (C). Total RNA was extracted from the various EC types and reverse transcribed. The PCR reaction was conducted with a 5′ primer specific for each ECE-1 isoform and a common reverse primer (see Table 1). (D) Amplification of either the ECE-1 full length (full) or the SV isoform separately was carried out using primers described in Table 1. Inverse images of ethidium bromide–stained agarose gels.

A schematic representation of ECE-1 gene structure and its different mRNAs. The ECE-1 gene structure showing the first alternative (1c, 1b, 2, and 3) exons and their promoters (p). Exons 4–19, common to all isoforms, which encode the major part of the ECE-1 cDNA, are not represented at the same scale. Exons are numbered according to Valdenaire et al. (18). Exons of the four different full-length ECE-1 mRNAs, together with those of the spliced variants mRNAs, are depicted at the bottom.

Detection of ECE-1 proteins in HEK-293 cells stably expressing bECE-1d, SV, and SVcut plasmids. Total proteins were extracted in lysis buffer and processed as detailed in Materials and Methods. Twenty-five micrograms of each cell extract was separated by sodium dodecylsulfate polyacrylamide gel electrophoresis under reducing conditions. Proteins were detected by Western blots using a C-terminal specific antibody (A) and a specific N-terminal antibody (B). NT, nontransfected cells.
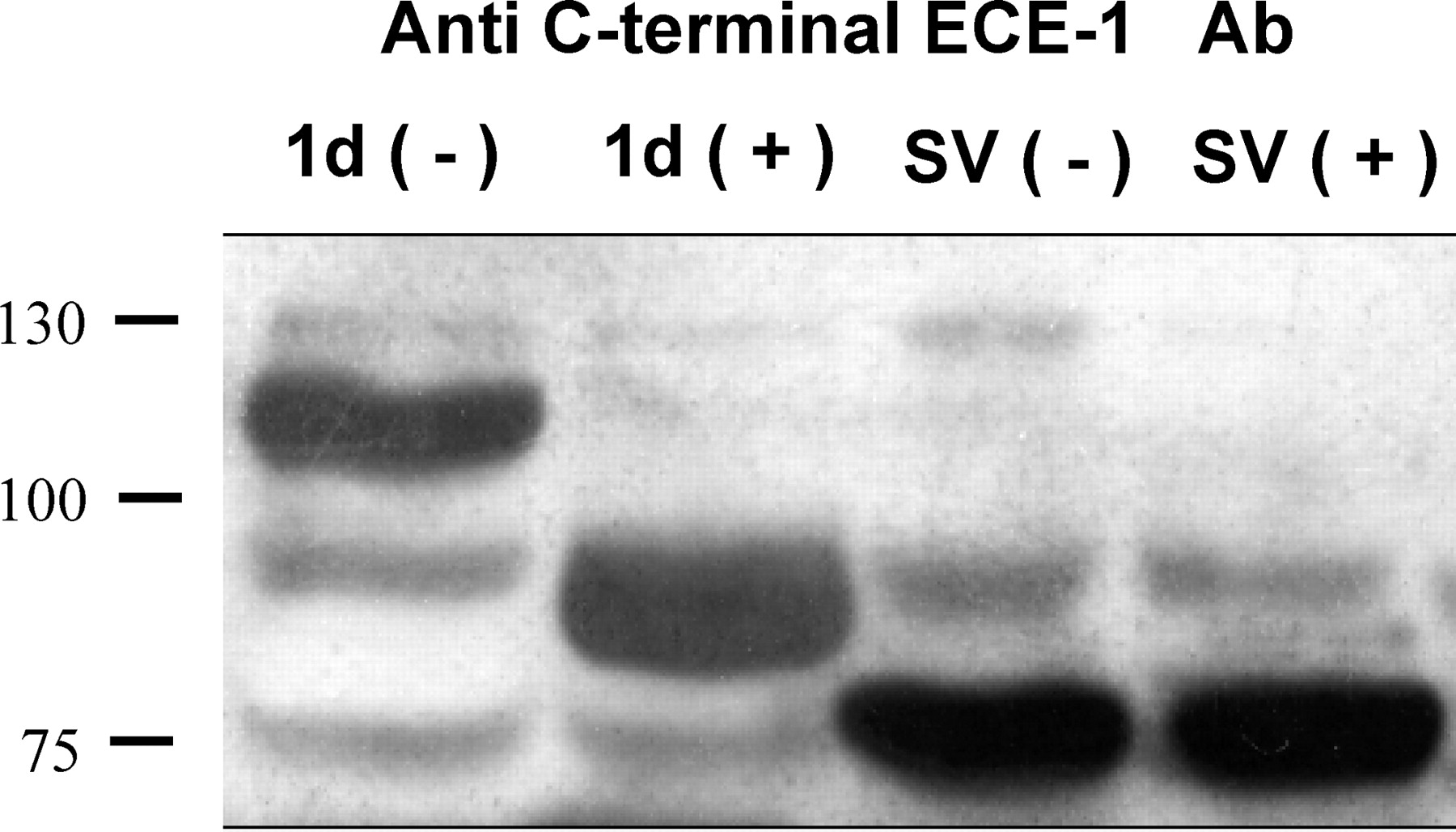
Deglycosylation of ECE-1d and ECE-1sv proteins. Recombinant proteins were incubated at 37°C for 1 hr without enzyme (−) or treated with 1500 U of N-glycosidase F (+), under reducing conditions and analyzed by Western blot using a C-terminal–specific antibody.
Footnotes
This work was supported by a grant from the Israel Science Foundation (ISF 0396189).