
Abstract
Endothelin (ET) levels are elevated in congestive heart failure secondary to myocardial infarction (MI) and correlate well with the severity of pulmonary hypertension (PH), suggesting that the ET peptide could contribute to the pathophysiology of venous PH. Alterations of pulmonary vasoreactivity to ET after MI and the respective roles of the ETA and ETB receptors (ETA-R and ETB-R) have never been evaluated, to our knowledge. MI was induced in rats. Three weeks later, small pulmonary resistance arteries were mounted on a microvascular myograph. Cumulative concentration-response curves to ET-1 and sarafo-toxin 6c (S6c) were performed. Response to ET was also assessed in the presence of ET-R antagonists. Heterodimeriza-tion of receptors was evaluated by immunoprecipitation of the ETB-R, followed by western blotting for the expression of the ETA-R. Maximal vasoconstriction and sensitivity to ET-1 were similar in sham and MI with values of 88 ± 3.9% and 80 ± 3.8%, respectively. The response to S6c was similarly less in both sham (67 ± 5.7%) and MI groups (60 ± 6.6%). When administered alone, the ETA-R antagonist (10 nM A-147627.1) and the ETB-R antagonist (1 μM A-192621.1) had no significant effect. However, their combination markedly reduced vaso-constriction (52 ± 5.3%; P < 0.001). The endothelial and medial distribution of ET-Rs was similar in sham and MI groups. In vitro studies demonstrated co-immunoprecipitation of the ETA-R and ETB-R. Vasoconstriction of isolated resistance pulmonary arteries to ET agonists is not altered after MI. Dual antagonism results in optimal blockade of vasoconstriction, possibly because the ETA-R and ETB-R can form functional heterodimers.
Introduction
Pulmonary venous hypertension is a frequent complication of congestive heart failure (CHF) that carries a poor prognosis. The endothelin (ET) system is activated in CHF of all etiologies, including myocardial infarction (MI), with plasma ET levels correlating especially well with the severity of secondary pulmonary hypertension (PH; Refs. 1–4). The pulmonary circulation is the primary site for both ET production and clearance in humans (5), and there is evidence for increase of both ET-1 expression (6, 7) and ET-converting enzyme (ECE) activity (8) in lung tissue of animal models of CHF.
In normal humans, approximately 50% of circulating ET-1 is extracted by the pulmonary circulation within a single transit time (5). This clearance is exclusively mediated by the endothelial ETB receptor (ETB-R; Ref. 9). Humans with CHF have reduced pulmonary ET-1 clearance that correlates well with the severity of PH (10). This is consistent with the demonstrated lower protein level of the ETB-R in the lung of CHF rats after MI (11). The specific contributions of the ETA-R and ETB-R on ET-induced pulmonary vascular reactivity after MI could, therefore, be modified and have significant physiopathologic implications.
Therefore, the aim of this study was to evaluate possible alterations of pulmonary vasoreactivity to ET-1 after MI and to characterize the respective roles of the ETA-R and ETB-R.
Materials and Methods
The animals research and ethics committee of the Montreal Heart Institute approved the study protocol. Experiments were conducted according to guidelines from the Canadian council for the care of laboratory animals. Male Wistar rats (250–300 g) were used for this study. Animals were submitted to sham operation (n = 11) or to MI (n = 33) induced by ligation of the left anterior descending coronary artery, as previously described (12).
Isometric Recording of Tension of Isolated Microvessels.
Three weeks after MI, rats were anesthetized for hemodynamic measurements, followed by isolation of small pulmonary arteries, as previously described (13). For each vessel, the integrity of the endothelium was determined by testing the endothelium-dependent vaso-dilation to acetylcholine (100 μM). The maximal vaso-constriction to 127 mM KCl was also determined for each vessel. Preparations were subjected to a cumulative concentration-response curve for ET-1 (0.1 nM to 0.3 μM) and sarafotoxin 6c (S6c; 0.1 nM to 0.3 μM). The ET-1 concentration-response curve was assessed in the presence of varying concentrations of an ETA-R antagonist (A-147627.1; 10 nM, 100 nM, 1 μM, and 10 μM), an ETB-R antagonist (A-192621.1; 1 μM, 10 μM, and 100 μM), or a combination of both antagonists.
Immunohistology of ET-Rs in Small Pulmonary Arteries.
Immunofluorescence and confocal imaging were performed as recently described in detail (13). ETA-R antibody (ETA, rabbit; Alomone, Jerusalem, Israel) and ETB-R antibody (ETB, rabbit; Alomone) were incubated respectively with a mouse antibody to α–smooth-muscle actin (Sigma Chemical Co., St. Louis, MO). The secondary antibodies, donkey anti-rabbit Alexa 555 (Molecular Probes, Eugene, OR) and donkey anti-mouse Alexa 647 (Molecular Probes), were diluted in their respective antibody diluents and applied. To quantify the fluorescence intensity of ETA-R and ETB-R, we used internal elastic lamina (IEL) and external elastic lamina (EEL) autofluorescence to identify the limits of the media and of the endothelium.
Confocal Imaging, Deconvolution, and Fluorescence Quantification.
Slides were analyzed using a Zeiss LSM 510 confocal microscope. We used a plan ApoChromat ×63/1.4 numerical aperature oil differential interference contrast objective. HeNel (543 nm) and HeNe2 (633 nm) lasers were used for excitation of the anti-rabbit Alexa 555 and anti-mouse Alexa 647 antibodies, respectively. IEL and EEL autofluorescence was obtained with the argon laser line (488 nm) and collected between 505 and 530 nm. Z-stacks of each tissues were performed and images were taken at every 0.16 μm (top to bottom) to respect the Nyquist criteria in z sampling. Z-stacks were deconvolved using the maximum likelihood estimation algorithm of the Huygens Pro software (version 2.4.1; Scientific Volume Imaging, Alexanderlaan, the Nether-lands). Transparent projections (in face view) were applied to each z-stack using the Projection tool of the LSM 510 software. Images were saved in TIFF file format. To quantify the fluorescence intensity of ETA-R and ETB-R, we used IEL and EEL autofluorescence to identify the limits of the media and the endothelium. Using the “close free shape curve” tool of the LSM 510 image software, we can isolate the endothelium or the media by masking the remaining of the image. Mean fluorescence intensity (MFI) was calculated over the nonmasked region by the LSM 510 software. This operation was executed at every fifth image of each z-stack. The MFI of all of the images in a z-stack were averaged.
Co-Immunoprecipitation.
Small intralobar pulmonary arteries from the pulmonary right inferior lobe were obtained and pooled (n = 24 arteries) for each experimental condition. Standard protein extraction was assessed using a lysis buffer with the following composition: 50 mM Tris-HCl, pH 7.5; 20 mM β-glycerophosphate; 20 mM sodium fluoride; 5 mM EDTA; 10 mM EGTA; 1 mM Na3VO4; 1% v/v triton; and protease inhibitor cocktail: 1 μM micro-cystein, 5 mM dithiothreitol, 10 μg/ml leuptine, 0.5 mM phenylmethylsulfonyl fluoride and 10 mM benzamidine; followed by protein quantification using the Bradford technique. A total of 100 μg protein from each sample was brought up to a volume of 100 μl with lysis buffer, and 1 μl of ETB-R antibody (Biogenesis, Hornby, Ontario, Canada) or of ETA-R antibody (Abcam, Cambridge, MA) was added. The sample was incubated at 4°C for 2 hrs, with agitation. While the samples were incubating with the first antibody, a washing buffer containing 1.5 mM Tris and 10% Triton X-100 in H2O was prepared. Three washes of agarose beads (Protein A/G PLUS-Agarose; Santa Cruz Biotechnol-ogy, Santa Cruz, CA) were carried out before the beads were incubated with each sample (20 μl/sample) at 4°C for 1 hr, with agitation. The samples were then washed thrice before the addition of sample buffer and preparation for immunoblotting.
Immunoblotting for ETA-R.
Proteins were separated on 10–20% (w/v) acrylamide gradient sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE). After SDS-PAGE, samples were transferred at 100 V and 4°C for 90 mins onto nitrocellulose membranes, in a buffer containing 25 mM Tris base, 192 mM glycine, and 5% methanol. Membranes were blocked for 2 hrs using a solution of 5% skimmed milk powder (Sigma) in 25 mM Tris-HCl, pH 7.5; 150 mM NaCl; and 0.05% Tween 20 (TBST). Membranes were incubated overnight at 4°C with primary antibodies (ETA-R; Abcam), diluted 1:1000 in 5% milk in TBST, washed with TBST (3 × 10 mins), reblocked with 5% nonfat milk in TBST (1 × 10 mins), and incubated for 2 hrs with the appropriate horseradish peroxidase–conjugated secondary antibody (anti-rabbit; Sigma) diluted in 1:20,000 in 5% nonfat milk powder. After extensive washing with TBST, immunoreactive bands were visualized by enhanced chemiluminescence (Renaissance Plus; Perkin-Elmer Life Sciences, Wellesley, MA) according to the manufacturer’s instructions, using Bio-Max film. The ETA-R (ab1919, rat ETA-R amino acids 31–45, SSHVEDFTPFPG-TEF; Abcam) and ETB-R (4113–3059, rat ETB-R amino acids 405–417, QTFEEKQSLEEKQ; Biogenesis Ltd.) antibodies were both raised against different and specific targets. We have tested the cross-reactivity of the ETA-R antibody in our endothelial preparation. The ETA-R antibody did not recognize the ETB-R of the endothelial preparation.
Study Drugs.
The ET-1 (American Peptide, Sunny-vale, CA) and S6c (American Peptide) agonists were used. The ETA-R antagonist, A-147627.1, and the ETB-R antagonist, A-192621.1, were kindly provided by Abbott Laboratories (Abbott Park, IL).
Statistical Analysis.
All values are expressed as mean ± SEM. Differences between the sham and MI groups for morphometric and hemodynamic parameters were analyzed by a two-tailed unpaired t test. For each pharmacologic condition on each isolated artery, the isometric recording of the concentration-response curves was fitted using a five-parameter logistic fit to determine the maximal responses and the median effective concentration (EC50) values. For these parameters, the differences between groups were evaluated with an unpaired Student’s t test. Statistical significance was assumed for P < 0.05.
Results
Three weeks after surgery, the infarct animals developed CHF with secondary venous PH, as evidenced by higher left ventricular end diastolic pressures (15 ± 2 mm Hg vs. 3 ± 1 mm Hg; P < 0.01) and right ventricular systolic pressure (38 ± 2 mm Hg vs. 29 ± 1 mm Hg; P < 0.05).
Reactivity of Small Pulmonary Arteries.
In isolated arteries, the maximal vasoconstriction and sensitivity induced by ET-1 were similar in sham and MI groups, with maximal (Emax) values of 88% ± 3.9% and 80% ± 3.8%, respectively (Fig. 1A). The response to S6c was similarly less in both shams (67% ± 5.7%) and MI groups (60% ± 6.6%; Fig. 1B). In the MI group, the ETA-R antagonist mildly reduced ET-1–induced pulmonary vaso-constriction, but this did not reach statistical significance. The ETB-R antagonist reduced vasoconstriction markedly, but only at the very high dose of 100 μM. To explore potential receptor interactions, experiments were performed by combining noneffective doses of these selective antagonists. The results are presented in Figure 2. In the MI groups, both the ETA-R antagonist (10 nM; Emax, 81 ± 2.5) and the ETB-R antagonist alone (1 μM; Emax, 88 ± 2.2) mildly reduced ET-1 vasoconstriction without affecting EC50 values (Fig. 2). The combination of both antagonists in sham (34 ± 9.0; P < 0.001; Fig. 2A) and MI groups (52% ± 5.3%; P < 0.001; Fig. 2B), however, greatly reduced Emax, with the EC50 becoming immeasurable.
Expression of ET-R in Small Pulmonary Arteries.
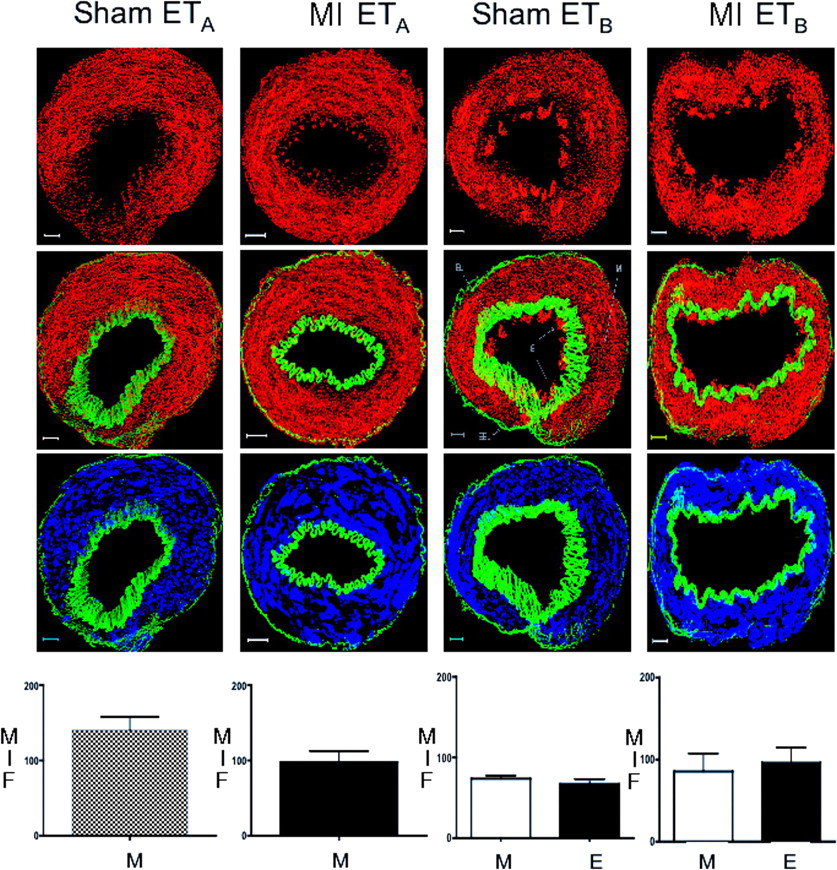
Examples of composite z-stack images obtained with the ETA-R and ETB-R antibodies and antibody to smooth-muscle actin are shown in Figure 3. Autofluorescence of IEL and EEL enables easy demarcation of the endothelium. As expected, the ETB-R was present on both the endothelium and media of pulmonary resistance arteries, whereas ETA-R was only present on the media. Fluorescence intensity revealed no difference between the ETA-R from MI lung preparations compared with sham preparations. Moreover, there were also no differences between the intensity of endothelial and smooth muscle ETB-R of MI lung preparations compared with sham preparations.
ETA-R Expression in Pulmonary Arteries After Immunoprecipitation of ET-Rs.
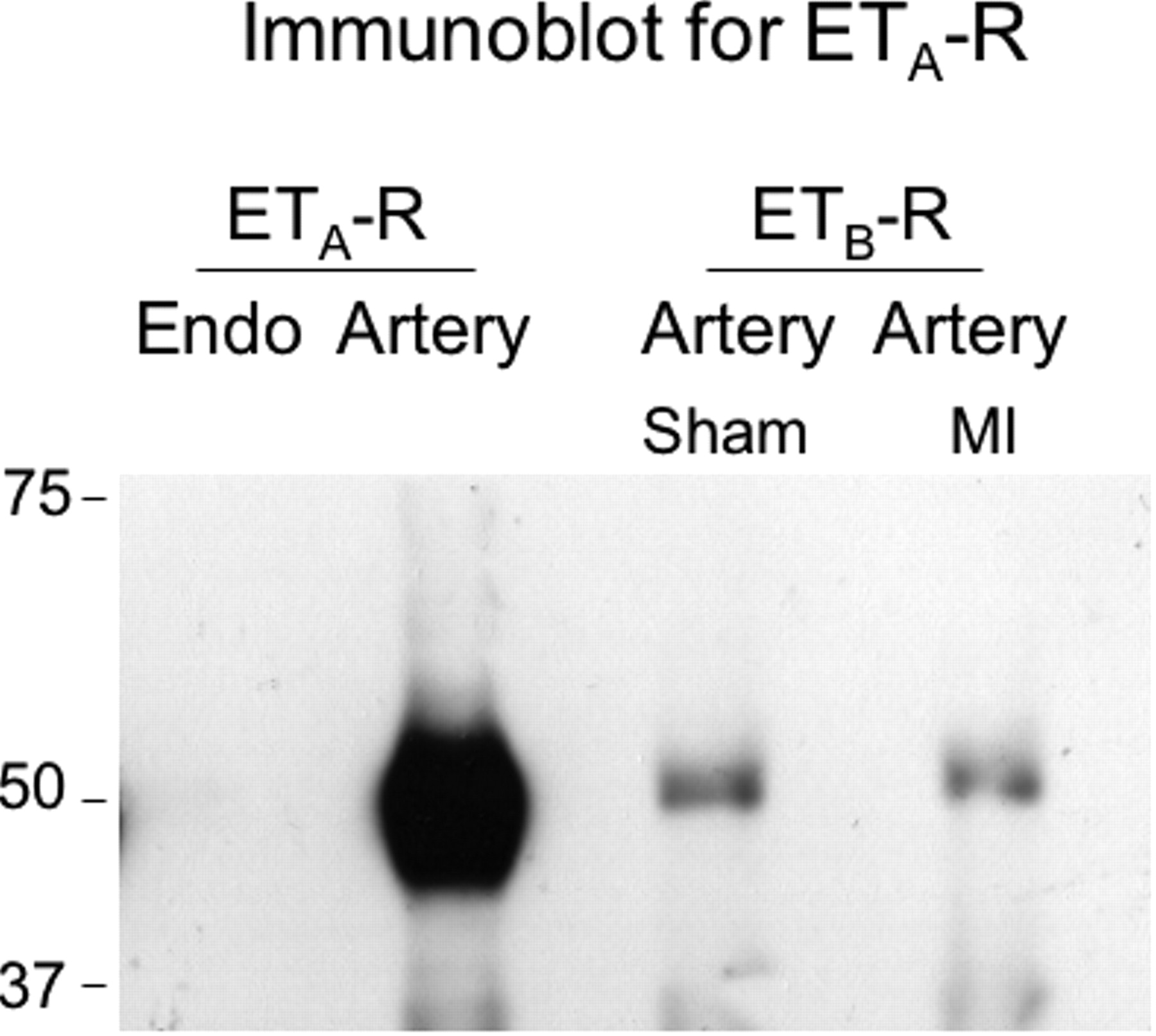
This experiment was undertaken to determine whether ET-R heterodimers were possible formed in the pulmonary circulation. After the ETA-R was immunoprecipitated, the immunoblot for ETA-R expression revealed a large and elongated band, indicative of the abundance of receptor protein (Fig. 4). When the ETB-R was immunoprecipitated, a narrow band for the putative ETA-R was expressed at 49.69 kDa, demonstrating that the ETA-R co-immunoprecipitated with the ETB-R. A negative-control experiment, carried out in aortic porcine endothelial cells lacking the ETA-R, confirmed the absence of detectable ETA-R after immunoprecipitation of the ETB-R. We additionally excluded the possibility that the observed band for the ETA-R could represent the heavy chain of the immunoprecipitation antibody, and again obtained no detectable band (data not shown).
Discussion
This study was designed to evaluate possible alterations of ET-1 vasoreactivity in pulmonary resistance arteries after MI. The results demonstrate that there is no modification of the response to ET-1 in this condition. It also demonstrates that selective stimulation of the ETB-R with S6c substantially contributes to vasoconstriction in both sham and MI animals. Consistent with these results, we demonstrate a lack of modification in ETA-R and ETB-R distributions in the lung circulation of the MI group. Finally, combined ETA-R and ETB-R blockade was necessary to obtain maximal inhibition of ET-1–induced vasoconstriction. A possible explanation for this finding was explored with the demonstration that ET-Rs can exist as heterodimers in both sham and MI rats.
It is well established that chronic stimulation of G protein–coupled receptors (GPCR) can result in their desensitization and downregulation. Our findings demonstrate that, in the case of ET, the documented activation of this system in CHF, with increased lung ECE activity and ET-1 levels (6–8) is, therefore, not associated with reduced ET-1–induced pulmonary vasoconstriction. This would suggest a lack of desensitization and/or downregulation of ET-Rs in small pulmonary arteries after MI. It is thus plausible that the good correlation between ET levels in CHF and the severity of associated pulmonary venous hypertension (1–4) may represent a cause-to-effect relationship.
Using immunohistology, we found that the relative distribution of ETA-R and ETB-R in small pulmonary arteries was not modified in the MI group. More specifically, we did not observe a modification in the proportion of endothelial ETB-Rs. In the same model, others have previously demonstrated a reduction of ETB-R messenger RNA expression and protein level in whole lung tissues (11). We have also previously observed a reduction of circulating ET-1 clearance in the lungs from this CHF model (14), as well as in humans with CHF (10), and interpreted these results as evidence of endothelial ETB-R desensitization and/or downregulation. A reduction of ETB-R density at sites other than the small resistance vessels evaluated in the present study could conciliate these apparently discordant findings.
More importantly, our evaluation of ET-1–induced vasoconstriction, in the presence of selective ET-R antagonists, confirms that dual blockade is necessary to obtain maximal inhibition. Indeed, both selective antagonists alone had little effect, except at the very elevated concentration of the ETB-R antagonist (100 μM), at which point, loss of selectivity probably occurred. When we combined noneffective concentrations of both selective ET-Rs antagonists, however, we observed a marked reduction in Emax. Consistent findings were previously observed in human resistance pulmonary arteries in which optimal blockade was achieved by dual inhibition of ET-Rs (15, 16).
Both ETA-R and ETB-R belong to the GPCR family. It has been firmly established that GPCR can form dimers or even higher-structure oligomers (17). The formation of heterodimers could modulate receptor function by regulating ligand binding and signaling, as well as receptor-trafficking properties. The heterodimerization could alter how a receptor functionally responds to a ligand, such that the antagonist of one receptor could then positively augment the action of the agonist of the associated receptor (18). Moreover, there has been new evidence that the ETA-R and ETB-R could form constitutive heterodimers (19). To further investigate the ETA-R and ETB-R functioning, we therefore, evaluated whether ET-Rs could exist as heterodimers in the pulmonary circulation. Immunoprecipitation of ETB-R and Western blotting for the expression of ETA-R confirmed that the receptors could form heterodimers in small pulmonary arteries. The functional implications of ET-R heterodimers in the pulmonary circulation remains highly speculative at this point, but our findings could partly be explained by the fact that only one of the two dimers could still induce vasoconstriction through compensation and signaling by the dimer not targeted by the antagonist. This is supported by the more complete inhibition of the ET response by a combination of both antagonists. Furthermore, our results suggest that dimerization of ET-Rs and the functional importance of dimerization is not modified after MI. Another possible, simpler explanation for our findings is that activation of either coexisting ET-R subtype alone can elicit maximal contraction and, thus, complete blockade of one type, leaving open activation of the other to induce maximal response.
In conclusion, we found that the vasoconstriction of isolated resistance pulmonary arteries to ET agonists is not altered after MI. ET-1–induced pulmonary vasoconstriction, in part, is mediated by ETB-R, and dual blockade is necessary for optimal inhibition of ET-1–induced vaso-constriction. The ET-Rs can exist as heterodimers in pulmonary arteries of both sham and MI rats, and this may have pharmacologic importance.
ET-1 (A) and S6c (B) induced vasoconstriction of pulmonary resistance arteries in
sham and MI rats. #P < 0.05; &P < 0.01
versus ET-1. Effect of selective and combined ETA-R and ETB-R blockade on
ET-1–induced vasoconstriction of pulmonary resistance arteries from (A) sham and (B)
MI rats. *P < 0.001 versus control. Distribution and quantification of ETA-R and ETB-R by
immunohistology of small pulmonary resistance arteries. The pictures represent
examples of composite z stacks that were deconvolved to measure MFI.
The first line displays the fluorescence for ETA and ETB from
left to right in both the sham and MI preparations (all in red). The second line
displays the same components, but with the addition of the IEL and EEL (in green),
which enables easy demarcation of the endothelium (E) from the media (M). The third
line displays the fluorescence for smooth-muscle actin (in blue), which is limited to
the media and colocalizes with the ETA-R (on the left) and the
ETB-R (on the right). The computed MFIs are presented in the bar
graph. Heterodimerization of ET-Rs in rat pulmonary resistance arteries. Immunoblotting for
ETA-R was performed after immunopre-cipitation of the ETA-R or
the ETB-R. Endothelial cells (endo, left lane) were used as a negative
control. Immunoprecipitation of the ETA-R in pulmonary artery (second lane)
was used as a positive control. The results confirm co-immunoprecipitation of the
ETA-R with the ETB-R in resistance pulmonary arteries of sham
and MI rats (right lanes).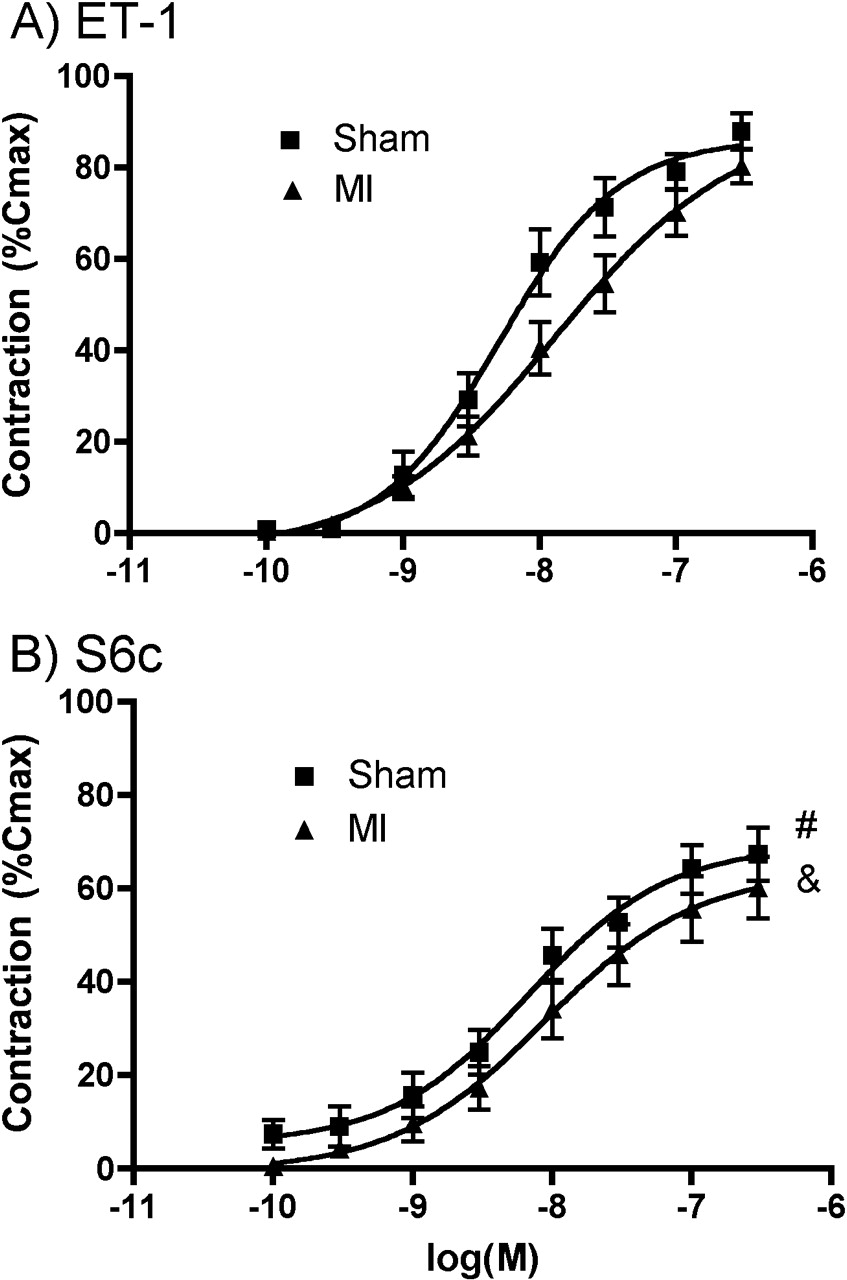

Footnotes
This work was supported by the Canadian Institutes of Health Research and the Fondation de l’Institut de Cardiologie de Montréal. Dr. Dupuis retains a national researcher scholarship from the Fonds de la recherche en santé du Québec. Dr. Thorin is a senior scholar from the Fonds de la recherche en santé du Québec.