
Abstract
Endothelin (ET)-1 is an autocrine/paracrine growth factor or an antiapoptotic factor in human cancers, and blockade of ET-1 receptors can sensitize human tumor cells to apoptosis. The role of the ET-1 axis in the proliferation and/or apoptosis of melanoma cells and in their response to the alkylating agent, dacarbazine (DTIC), used in clinical treatment of human melanoma were investigated in five human melanoma cell lines obtained form surgical resection specimens. Melanoma cells expressed the messenger RNAs (mRNAs) for the components of the ET-1 axis. ET-1 binding was mediated by ETB but was inhomogeneous among melanoma cells. Exogenous ET-1 did not induce human melanoma cell proliferation. Bosentan, a dual ETA/B-receptor antagonist, decreased melanoma cell viability and DNA synthesis and induced melanoma cell apoptosis in defined human melanoma cells. Bosentan potentiated Fas ligand–induced apoptosis only in one melanoma cell line. Variants of ET B were determined using reverse transcriptase (RT) polymerase chain reaction (PCR) and primers spanning the whole sequence of the ET B gene. ET B variants were demonstrated only in one of the five cell lines, corresponding to the absence of ET-1 binding by these cells. Bosentan did not inhibit the effects of alkylating agents, and the effects of bosentan and alkylating agents were additive in melanoma cells. In conclusion, exogenous ET-1 is not a growth factor for human melanoma cells, but blockade of ET receptors decreases proliferation, induces apoptosis, and potentiates the effects of anticancer agents in defined melanoma cells, suggesting that combination therapy of ET-receptor antagonists with alkylating agents may improve their efficacy.
Introduction
Endothelin (ET)-1 overexpression in human cancer has been associated with cancer progression. The effects of ET-1 on mammalian cells are mediated by two distinct subtypes of G-protein–coupled receptors, ETA and ETB (1, 2), which are frequently overexpressed in cancer and stimulate several intracellular signaling pathways mediating a variety of physiologic functions, including cell growth and death. Antagonists to ET-receptors, which were initially developed for cardiovascular diseases, may also be useful in clinical treatment of human cancers.
The control of tumor cell proliferation and apoptosis is involved in the mechanisms of many chemotherapeutic agents used to treat cancer. In a pilot study, the efficacy of bosentan, a dual ETA/B-receptor antagonist, was evaluated in patients with advanced malignant melanoma, resulting in disease stabilization in a significant number of patients. The results also suggested that combination therapy would improve treatment efficacy. First-line chemotherapeutic treatment of human malignant melanoma includes the use of alkylating agents, such as dacarbazine (DTIC), in combination with other drugs. However, resistance to apoptosis and to inhibition of proliferation of cancer cells has been described, involving various mechanisms, and indicating that antiapoptotic pathways allow transformed cells to escape death. The resistance of cancer cells to alkylating agents involves the DNA repair enzyme, O6-methyl-guanine methyltransferase (MGMT/AGT), which is able to repair DNA alkylation. The MGMT/AGT promoter is frequently methylated, and the enzyme nonexpressed in human cancers (3, 4). We have previously shown in human glioblastoma and colon carcinoma cells that ET-1 is not a proliferation-inducing factor, but an antiapoptotic factor, and that antagonists of the ET receptors or inhibition of the endothelin-converting enzyme (ECE)-1 sensitized human tumor cells to Fas ligand (FasL)–mediated apoptosis (5–9). However, in these previous studies, not all cells responded to ET receptor blockade and/or were sensitized to apoptosis, suggesting that the combination therapeutic regimen may be a more efficient treatment than monotherapy with ET receptor antagonists alone. On the assumption that ET-1 might also be involved in resistance of human melanoma cells to apoptosis, we first studied the ET-1 system in five human melanoma cell lines and their response to ET-1, ETA/B antagonism, and FasL, then we evaluated the effects of combination therapies of alkylating agents and the dual ETA/B receptor antagonist, bosentan, in human melanoma cells expressing or not expressing MGMT/AGT, to determine whether the expression/promotor methylation of MGMT/AGT may be predictive of response to combination treatment.
Materials and Methods
Cells and Cell Treatment.
Human melanoma cell lines (10) were derived in Lausanne from surgical specimens, according to the Hospital Ethics Committee and patients’ agreement. Cells were grown in RPMI-1640 medium containing 4.5 g/l glucose, 10% fetal calf serum (FCS), and antibiotics (all from Gibco-BRL, Basel, Switzerland). For experiments, cells were grown in 48-well plates (Costar, Corning, NY) in the presence of FCS, and deprived of FCS for the time indicated. The cultures were exposed to bosentan (a kind gift of Actelion, Alschwill, Sitzerland), and/or the alkylating agent, DTIC-Dome/DTIC, stock (solution 30 mg/ml (165 mM in RPMI-1640, pH adjusted to 7.2 with carbonate solution), and either the MTT assay to determine the number of metabolically active cells, thymidine incorporation to determine DNA synthesis, or the quantification of DNA nucleosome fragments to determine apoptosis were performed (see “Apoptosis Evaluation” below).
Evaluation of ET-1 Binding to Cells.
Melanoma cells were grown to confluence in the presence of FCS. Medium was changed to fresh medium containing 10% FCS, and increasing concentration of either BQ123 (ETA selective) or BQ788 (ETB selective), both from Alexis Biochemicals (Lausen, Switzerland) or 200 nM ET-1 (Bachem, Bubendorf, Switzerland) plus 50 pM 125I–ET-1 (Anawa Trading, Wangen, Switzerland) were added. After 1 hr at room temperature, the cell layer was washed, extracted in 0.1% sodium dodecyl sulfate in 0.1 N NaOH, and the supernatants and cell extracts were counted in a gamma counter (Cobra, Packard, Foster City, CA).
Detection of Messenger RNAs (mRNAs) by Reverse Transcriptase (RT) Polymerase Chain Reaction (PCR).
Total RNA from confluent cells was prepared using Trizol reagent (Gibco-BRL), and RT-PCR for the ET-1 system was performed according to standard procedures and specific primers, as previously described, with preproET (PPET)-1 (341 base pairs [bp]), ECE-1 (622 bp), ET A (675 bp), and ET B (400 or 425 bp; Refs. 5, 8). To ensure the quality of the RNAs, amplification reactions were performed with pairs of primers specific for human glyceraldehyde-phosphate dehydrogenase (GAPDH, 456 bp; Ref. 5). To probe for potential ET B variants, primers were designed based on published information (11–14) or designed from the published sequence of ET B . Their sequence, characteristics, and location on ET B are shown in Table 1.
Evaluation of Cell Growth.
3,4,5-Dimethylthiazo-lyl-2,5-diphenyl tetrazolium (MTT) reduction to quantify metabolically active cells, and thymidine incorporation to assess DNA synthesis were performed, as previously described (5, 8, 9). Briefly, after treatment, cells were exposed to 0.25 mg/ml MTT (Sigma, Buchs, Switzerland) for 2 hrs, and the precipitated formazan was dissolved in 0.1 N HCl in isopropanol and quantified at 540 nm (iEMS, Labsystems; Bioconcept, Allschwil, Switzerland). Alternatively, after agent addition, 1 μCi/ml 3H-thymidine (Amersham Pharmacia, Dübendorf, Switzerland), was added for 3–24 hrs, and incorporation was quantified in a beta counter (Rackbeta, LKB, Wallac, Turku, Finland) after precipitation with 10% trichloracetic acid and solubilization in 0.1 N NaOH.
Bosentan and FasL-Induced Apoptosis.
The murine neuroblastoma cell line, Neuro-2A FasL, has been transfected to produce soluble murine FasL and was used as a source of active FasL, as previously described (6). Melanoma cells were grown to half confluence in RPMI-1640 supplemented with 10% FCS, the cells were deprived of FCS for 4 hrs, and then incubated for 24 hrs in the absence of FCS and with FasL (diluted 1:5) and/or bosentan (8 and 80 μM). Apoptosis was evaluated 24 hrs later (see “Apoptosis Evaluation” below).
Apoptosis Evaluation.
Apoptosis was quantified as previously described (5–8) using the Cell Death Detection ELISAPLUS kit (Roche, Rotkreuz, Switzerland), a photometric enzyme-linked immunoassay for quantitative in vitro determination of cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes), according to the supplier’s instructions. The apoptosis index was calculated as the ratio of absorbance of treated cells to the absorbance of untreated cells.
Determination of MGMT/AGT Promoter Methylation, mRNA, and Protein.
DNA was extracted from confluent layers of melanoma cells using the DNeasy tissue kit (Qiagen, Hombrechtikon, Switzerland), reacted with bisulfite using standard protocols, followed by purification using a QIAquick PCR purification kit (Qiagen), and the determination of promoter methylation was performed using primer for methylated and unmethylated selective promoter sites by PCR, as previously described (3, 4). MGMT/AGT mRNA and protein were determined by RT-PCR using the following primers: 5′-GACAAGGATTGTGAAATGAAACG-3′ and 5′-TCAGTTTCGGCCAGCAGGC-3′ and Western blotting using an anti-MGMT/AGT polyclonal antibody (SC-8825; Santa Cruz Biotechnologies, Santa Cruz, CA).
Calculations of Results.
Each experiment was repeated at least three times, unless otherwise stated. Means and standard deviation were calculated. Statistical significance was assessed using an unpaired two-tailed Student’s t test.
Results
The origin of Me190, Me191, Me237, Me275, and Me300 human melanoma cells and their expressions of the mRNAs of the ET-1 axis were determined by RT-PCR and are shown in Tables 1 and 2. The presence of functional cell membrane ET-1 receptors in all melanoma cells was evaluated using 125I–ET-1 binding (Fig. 1A). Two cell lines, Me275 (Fig. 1A) and Me300 (data not shown), bound ET-1 at high levels; two cell lines, Me190 and Me237, displayed only a low level of ET-1 binding; and ET-1 binding to Me191 cells was below detection limits (data not shown). Displacement studies by BQ123 (ETA selective) and BQ788 (ETB selective) demonstrated that, in all cells, ET-1 binding was exclusively mediated by ETB (Fig. 1A, Me275 cells). At very high concentration, BQ123 is no longer selective for ETA, and some antagonistic effects for ETB can be observed. Displacement constants were calculated to be approximately 50 pM, similar to previously published values.
In all serum-deprived melanoma cells, ET-1 did not induce cell proliferation as determined either by MTT reduction, which quantifies metabolically active cells; or 3H-thymidine incorporation, which quantifies DNA synthesis (Fig. 1B, Me275 cells). At 80 μM, bosentan, a dual ETA/B receptor antagonist, inhibited DNA synthesis in all melanoma cells (Fig. 1C). Furthermore, 80 μM bosentan statistically decreased the number of metabolically active cells after 24 hrs of exposure, with the exception of Me191 cells (Fig. 1D).
FasL alone induced apoptosis in Me237 and Me275 cells (Fig. 2). Bosentan induced apoptosis in Me190, Me191, and Me275 cells in a dose-dependent fashion, and potentiated FasL-induced apoptosis in Me275 cells (Fig. 2), whereas Me300 cells were resistant to all apoptotic stimuli. It had been previously shown that all cell lines expressed Fas (10; and unpublished results 1 ). Thus, the responses of melanoma cell lines to bosentan and/or FasL were heterogeneous.
D. Rimoldi, May 2002.
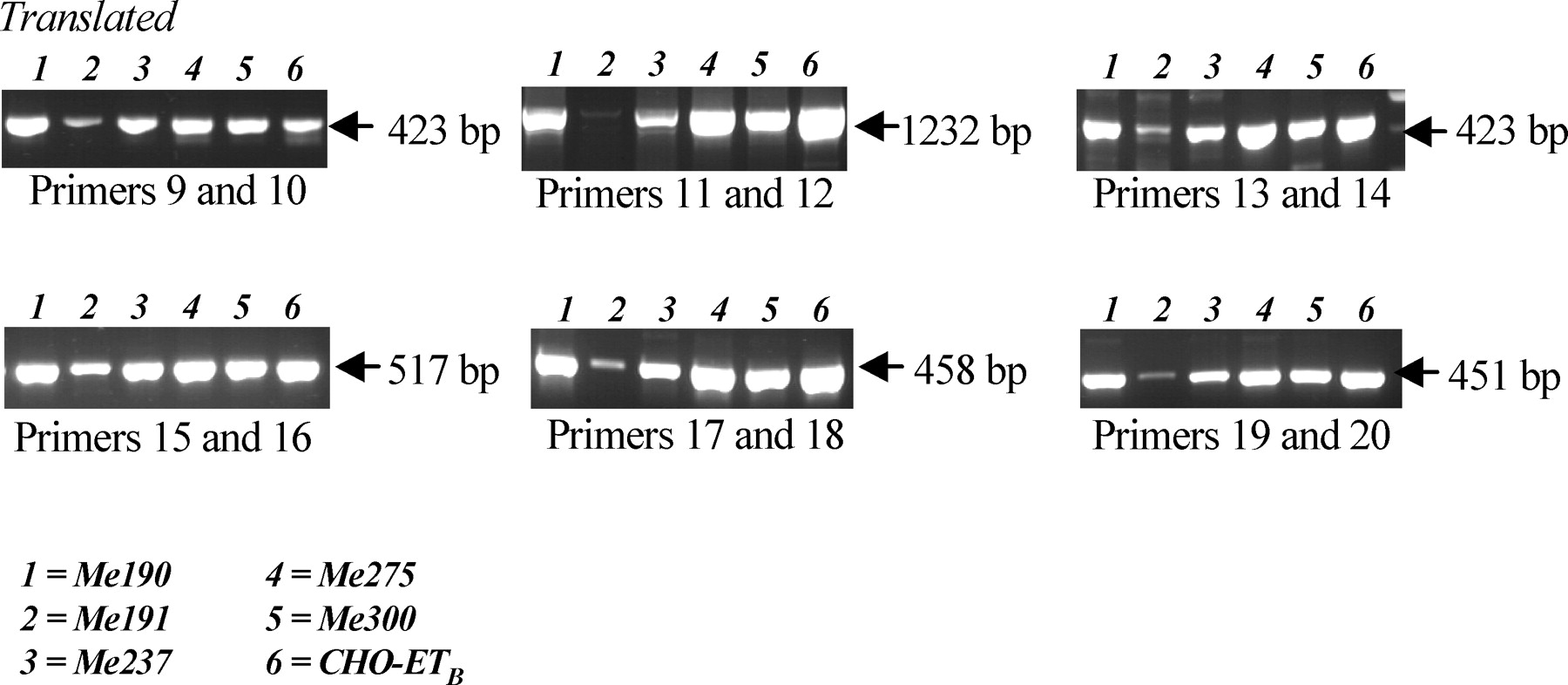
To determine whether alternative variants of ET B may be involved in the differences that we observed for ETB binding properties between cell lines and responses to bosentan, we performed RT-PCR for ET B using primers (Table 1) spanning all domains of the ET B sequence. As a positive control, we used Chinese hamster ovary (CHO) cells transfected with the coding sequence of ET B under a cytomegalovirus (CMV) promoter (a kind gift of Dr. F. Pinet, Collège de France, Paris). We detected variant forms of the ET B only in Me191 melanoma cells (Fig. 3), correlated with the absence of ET-1–binding capacity of these cells. The pattern of expression suggested that the N-terminal and C-terminal domains of the protein, but not its central core, would be defective.
Finally, we determined whether bosentan has the potential to interact with the effects of the alkylating agent, DTIC, used as chemotherapeutic agent to treat melanoma (Fig. 4). We did not observe inhibition of the effect of the alkylating agent by bosentan (either at 8 μM, 20 μM [data not shown], or 80 μM; Fig. 4A), and the bosentan and DTIC effects were rather additive (Fig. 4A) in the three melanoma cells that responded to bosentan for apoptosis, Me190, Me191, and Me275, as well as in Me237 (data not shown), suggesting that combination therapy may improve treatment outcome. To determine whether the expression of the enzyme MGMT/AGT, which is involved in the resistance to alkylation-inducing agents depending on its promoter methylation state, was involved or predictive to response, we assessed its promoter methylation, mRNA, and protein expression (Fig. 4B). No correlation between MGMT/AGT promoter methylation, and either mRNA or protein expression (Fig. 4B) and response to either bosentan or DTIC was found.
Discussion
Tumor progression is dependent on equilibrium between cell death–promoting and growth-promoting factors. ET-1 may be growth promoting in human ETA-expressing carcinomas (6–8, 15), although this latter effect was not generally observed in noncarcinoma ETB-expressing human glioblastoma cells (5), whereas in animal models of melanoma (16, 17), blockade of ETB slowed cancer progression. However, in these animal studies, either a short-time evaluation was performed (16), or the results showed that after an initial regression, colon carcinoma progression resumed (6). These results suggested that tumor cell selection for resistance to ET-receptors blockade arose. The present study was, therefore, undertaken to clarify the role of the ET axis in human melanoma cell proliferation and apoptosis.
Human melanoma cells expressed the mRNAs of the components of the ET axis, however, only ETB-mediated binding was observed, and the level of this binding was heterogeneous. In agreement with our present results, it has been previously shown (18, 19) that ETA is not expressed in melanoma cells, and that expression of ETB predominates, although it is variable. In addition, ETB was downregulated in melanoma compared with melanocytes, and ETB expression was enhanced in metastases compared with primary tumors. We did not observe such a relationship, because primary and metastatic melanoma cells exhibited low or high ETB-mediated ET-1 binding. Exogenous ET-1 was shown to be mitogenic for melanocytes, but not for melanoma cells (18, 19). In agreement with this previous observation, exogenous addition of ET-1 did not increase proliferation and growth of any of the melanoma cells evaluated, irrespective of ET-1 membrane binding properties. Bosentan and FasL induced apoptosis in three and two cell lines, respectively, and potentiation of FasL-induced apoptosis by bosentan was observed only in one (Me275 cells) out of the five human melanoma cells. Bosentan potential to induce apoptosis was not related to the ET-1 membrane binding properties of the cells. However, it was previously shown (20) that endogenous production of ET-1 or ET-3 by cells decreased the number of binding sites for exogenous ET-1 or ET-3, possibly by active endocytosis and targeting of ETB/ET-1 to the lysosomes (21). It has been shown (22) that ET-1 reduces the basic apoptosis rates via an ETB-mediated effect in melanocytes and melanoma and that ETB overexpression after transfection results in the restoration of the antiapoptotic response. Recently, it was demonstrated that 7-day exposure to the ETB-selective antagonist, BQ788, induced apoptosis more effectively in metastatic than primary melanoma cells (17), further suggesting that defined cell populations may be sensitive or resistant to ET receptor antagonisms. Our results support such a relationship using the dual ETA/B antagonist, bosentan, and FasL, and shorter exposure time of the cells. However, a high concentration of exogenous ET-1 has also been shown to induce ETB-mediated apoptosis in the human A375 melanoma cells (23, 24). In human glioblastoma cells, we also observed that ET-1 binding was exclusively mediated by ETB, and that ETB was a survival, but not a mitogenic factor, for glioblastoma cells (9); and that ET-1 at high concentrations induced apoptosis in human glioblastoma and colon carcinoma cells (8, 9). In glioblastoma cells, bosentan alone induced apoptosis and potentiated FasL-induced apoptosis in a subpopulation of cells. Therefore, ETB is linked to survival/antiapoptotic functions rather than to mitogenic activity in human melanoma cells, and the ET-1/ETB axis promotes melanoma progression by protecting cells against apoptosis. The exact molecular bases of these effects are not known. However, the response was heterogeneous, depending on the particular melanoma cell line evaluated. Similar observations were reported using human melanoma cells different from ours (16). Moreover, in the present study, as in our present and previous studies using human and rat cancer models (5, 6, 8), ETB antagonists inhibited growth and induced death of some, but not all, human tumor cells, but only at high (>40 μM) concentration of the antagonists, higher than the concentration necessary to displace ET-1 from its binding sites on membrane-expressed ETB.
Thus, blockade of ET-receptors was able to induce apoptosis only in a subpopulation of melanoma cells, and this effect was not correlated to the level of exogenous ET-1 binding to its membrane receptors(s), suggesting the presence of an alternative binding site for ET peptides and for receptor antagonists. In previous studies using colon carcinoma cells (6–8), we demonstrated biphasic apoptotic effects of ET-1 in the presence of bosentan, depending on the concentration of this peptide. This observation may suggest that membrane and intracellular receptors are involved in these effects. Pharmacologic studies using agonists and antagonists have also suggested the existence of additional ETB subtypes, and several alternative and splice variants of both ET A (25, 26) and ET B (27–29) receptors have been described, resulting either in in-frame addition or truncation of the N-terminal domain (the binding site for ET peptides), and normal functional properties but highly reduced cell-surface expression of ETB, although the C-terminal domain was not modified. The C-terminal domain is involved in lysosomal sorting of ETB (21). It has also been demonstrated that the 5′ flanking region of ETB contains numerous CpG sites, whose methylation decreased expression and resulted in the loss of functional coupling involving the C-terminal tail in prostate cancer (11–14). Therefore, using RT-PCR and primers spanning the sequence of ET B , we investigated whether variants of ET B can be found in human melanoma cells, which may explain the discrepancies observed between the cell response to bosentan and the binding properties for exogenous addition of ET-1. We found evidence for the absence, or only very low expression, of the ET B complete translated sequence only in Me191 cells. The pattern of ET B mRNA expression suggested that the N-terminal and C-terminal domains of the protein would be defective and unable to bind ET-1 in these cells.
Our results also suggested the development of resistance to the proapoptotic effects of antagonists of ET receptors in subpopulations of human melanoma cells. Therefore, we evaluated whether a chemotherapeutic agent used in clinics to treat melanoma, the alkylating agent DTIC, can potentiate bosentan effects. Combination of drugs in cancer may increase efficacy by two different ways: increasing sensitivity to one drug by the other, using similar intracellular signaling pathways in the same cells; or additive effects using different signaling pathways or different tumor clones. DTIC is a first-line chemotherapeutic agent for melanoma treatment; the effects of DTIC may be mediated by DNA synthesis inhibition as a purine analog, as a thiol reactive molecule, or as an alkylating agent. Our results did not reveal any reciprocal potentiation or inhibition between the alkylating agent and bosentan, but their effects were rather additive. Therapy tailored to the tumor characteristics is becoming an option in cancer; therefore, we determined whether the expression of MGMT/AGT, which is involved in alkylation-resistance mechanisms, may be an indicator for response to bosentan and/or DTIC. Our results demonstrated that this was not the case in human melanoma cells. Recently, we have shown that ECE-1 inhibitors inhibited DNA synthesis in human glioblastoma cells, and that exogenous addition of ET-1 did not counterbalance the growth inhibition elicited by ECE-1 inhibitors independently of the expression level of the functional cell membrane ET-1 receptor (9), suggesting that the combination of the blockade of big ET-1 activation and ET-1 receptors may be an option in cancer treatment.
In conclusion, we have shown that ET-1 was not a proliferation-inducing factor in human melanoma cells. Blockade of ET receptors by bosentan either directly induced apoptosis or was able to potentiate FasL-induced apoptosis in selected cells. However, the proapoptotic response of melanoma to bosentan and/or FasL was heterogeneous, suggesting that subpopulations of melanoma cells would escape apoptosis, and that a combination of treatments will be required. We show here that a combination of an antagonist of ET-receptors, such as bosentan, and alkylating agents, such as DTIC, represents such a possibility.
Sequence and Size of the Primers Used to Determine Variants of ET B

Characteristics and Expression of the ET-1 System of the Human Melanoma Cell Lines a
Comparison of the Effects of Bosentan Combined with the Alkylating Agent, DTIC, the Apoptosis Inducer, FasL, and the Expression of MGMT/AGT on the Growth of Human Melanoma Cells a
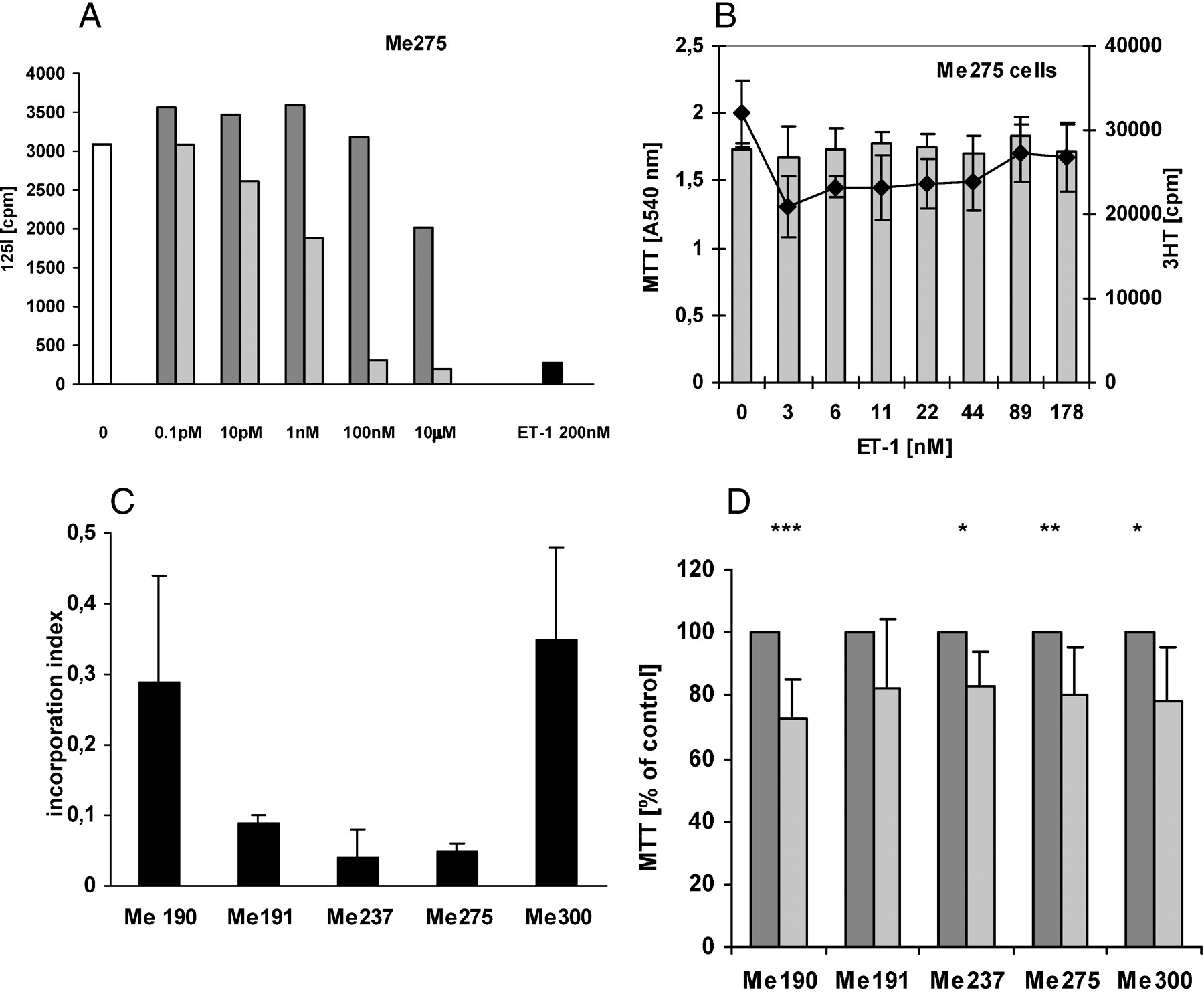
The function of the ET-1 axis in the growth of human melanoma cells. (A) ET-1 binding to melanoma cells is mediated by ETB. Confluent Me 275 cell layers were exposed to either no effector (white bar), 200 nM ET-1 (black bar), or increasing concentrations of either the selective ETA antagonist, BQ123 (dark gray bar), or the selective ETB antagonist, BQ788 (light gray bars), then radioactive ET-1 was added and the ET-1 binding to the cells was determined. The 50% inhibitory concentration was evaluated to be approximately 50 pM. (B) ET-1 does not induce melanoma cell growth. Me275 melanoma cells were grown to half-confluence for 24 hrs, deprived of FCS for 24 hrs, increasing concentrations of ET-1 were added for 24 hrs, and either MTT assay or 3H-thymidine incorporation was performed for the last 2 hrs. Gray bars: MTT;♦: 3H-thymidine incorporation. (C) Bosentan inhibits the growth of melanoma cells. Me190, Me191, Me275, Me237, and Me300 human melanoma cells were grown to half confluence for 24 hrs, deprived of FCS for 8 hrs, increasing concentrations of bosentan were added for 16 hrs, and 3H-thymidine incorporation was performed for the last 2 hrs. (D) Bosentan decreases the number of metabolically active cells. Cells were grown for 72 hrs, deprived of FCS for 24 hrs, then 80 μM bosentan was added for 24 hrs, and the MTT assay was performed for the last 2 hrs. Dark gray bars: no bosentan; light gray bars: with bosentan.
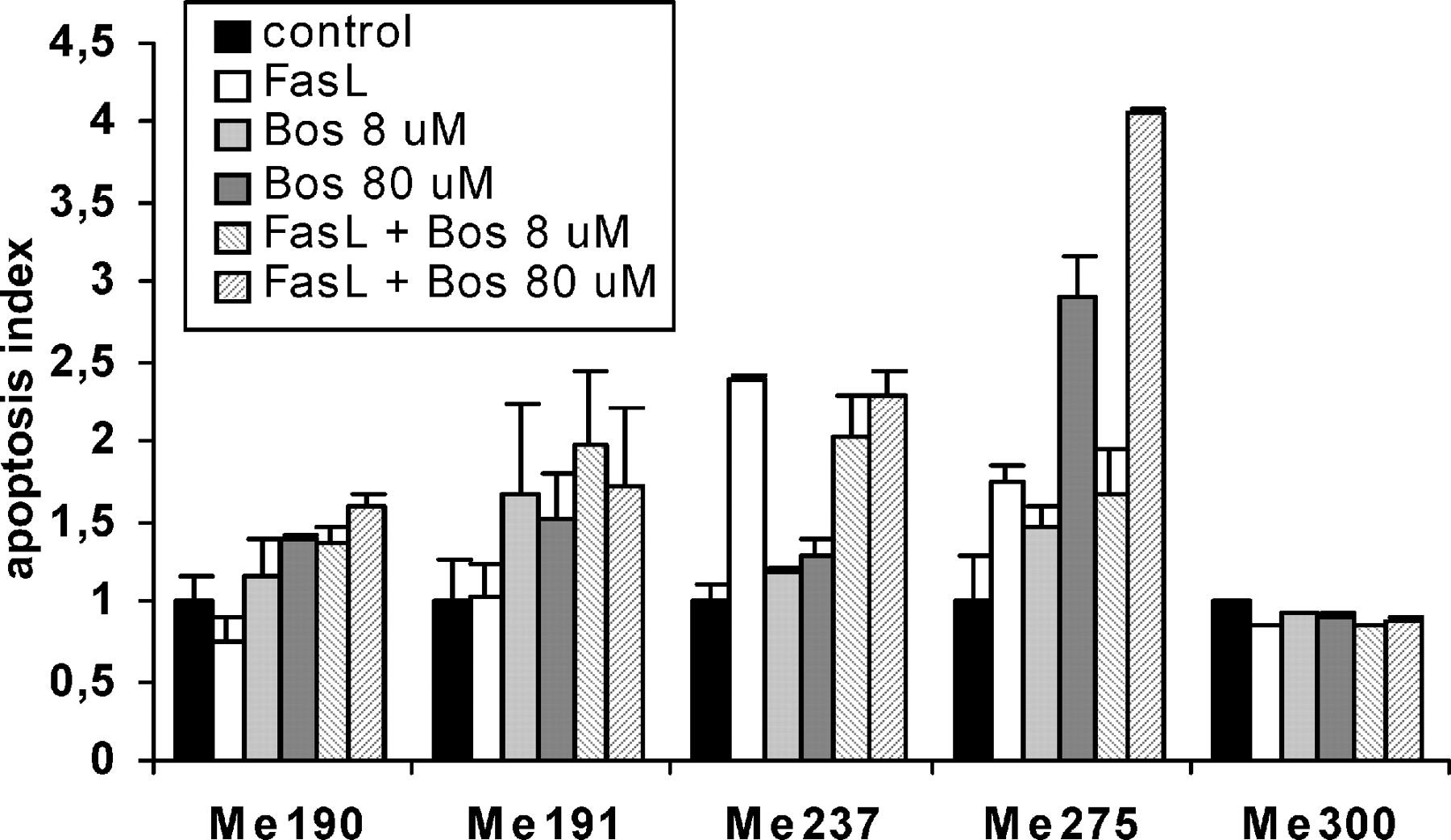
Bosentan and/or FasL induce apoptosis of melanoma cells. Me190, Me191, Me 275, Me237, and Me300 human melanoma cells were grown for 72 hrs, deprived of FCS for 4 hrs, then either FasL, bosentan (8 μM or 80 μM), or a combination of both was added to melanoma cells in the absence of FCS. Apoptosis was evaluated after 24 hrs of incubation. Apoptosis index = 1 is defined as the apoptosis measured in the absence of all effectors. Means ± SD of quadruplicate wells were calculated.
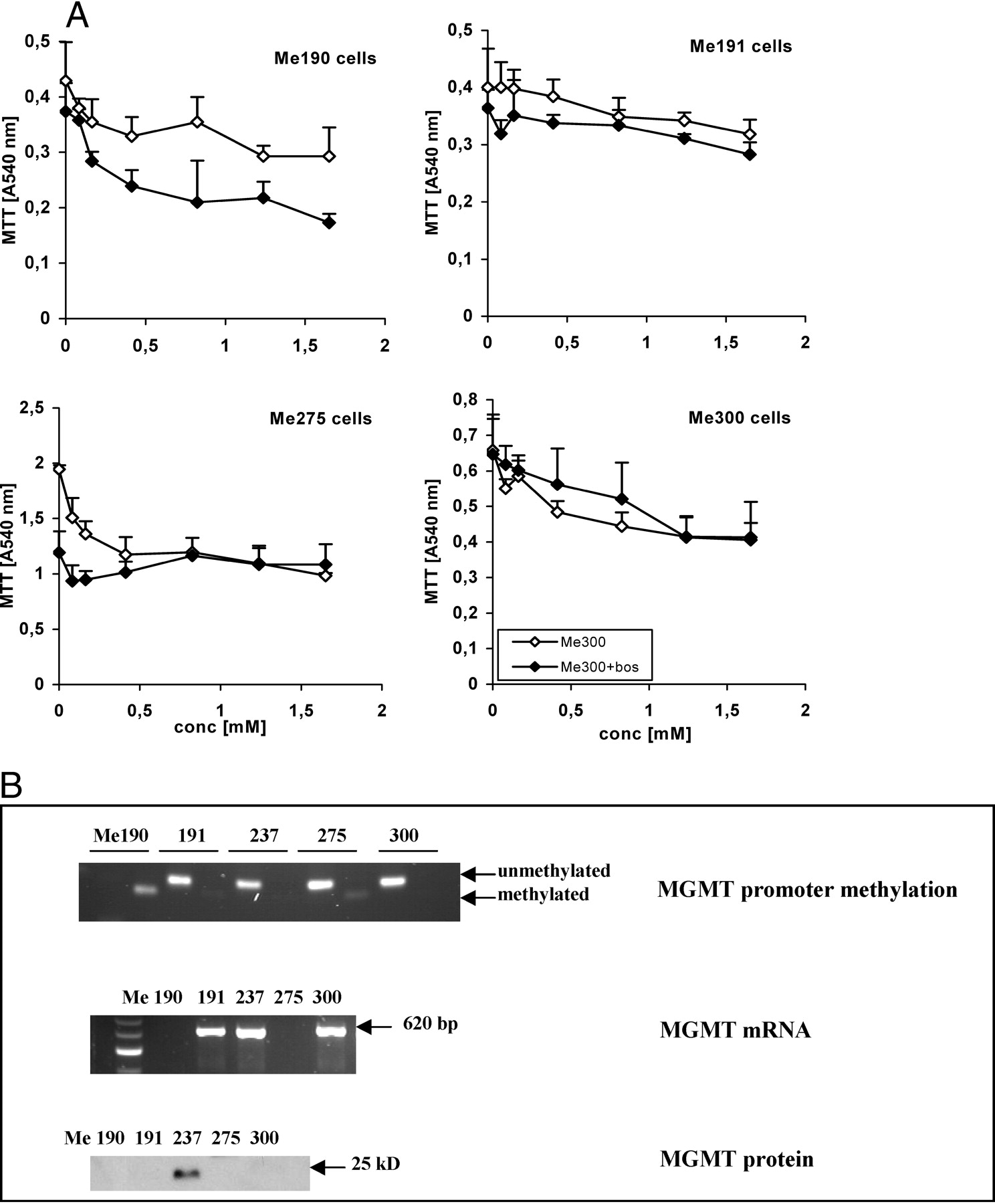
MGMT expression in melanoma cells and response to the alkylating agent, DTIC, and bosentan. (A) Me190, Me191, Me 275, and Me300 human melanoma cells were grown for 72 hrs, deprived of FCS for 24 hrs, and 80 μM of bosentan and increasing concentrations of DTIC were added to melanoma cells in the absence of FCS for 24 hrs. The MTT assay was performed for the last 2 hrs. (B) MGMT/AGT promoter methylation (MGMT promoter methylation) was determined by PCR after a bisulfite reaction of DNA extracted from melanoma cell DNA; MGMT/AGT mRNA (MGMT RNA) and protein (MGMT protein) were determined by RT-PCR and Western blotting, respectively.
Footnotes
This work was supported by grant from the Swiss League and Research against Cancer (SKL 353-9-1996, KFS 947-09-1999, KFS 1070-09-2000, and KLS-01308-02-2003), and the Swiss National Foundation for Scientific Research (grant 3200-064907.01).
Acknowledgements
We thank Ms. S. Gros, S. Trepey, P. Fioroni, and B. Carnal for excellent technical assistance; Dr. D. Rimoldi, Ludwig Institute of Cancer Research, Lausanne for the generous gift of melanoma cells and very helpful comments and suggestions; Dr. A. Fontana and Dr. F. Pinet for the kind gift of FasL-producing cells and ETB-transfected CHO cells, respectively; J.-D. Aubert for helpful discussions and suggestions, and M. Clozel from Actelion (Basel) for providing bosentan and for very helpful comments.