
Abstract
In addition to causing overt nociception, intraplantar (ipl) endothelin (ET)-1 injection into the rat hind paw induces hyperalgesia to mechanical stimuli, mediated via local ETB receptors coupled to protein kinase (PK) C, but not PKA. The present study further examines the intracellular signaling mechanisms underlying this effect of ET-1. ET-1 (30 pmol) or phospate-buffered saline (PBS) was injected ipl in rats and the threshold of responsiveness to mechanical stimulation was assessed repeatedly each hour up to 8 hrs and 24 hrs, using the dynamic plantar aesthesiometer test, which detects the minimal pressure required to evoke paw withdrawal. Different groups were treated, 15 mins before ET-1 administration, with ipsilateral injection of selective inhibitors of either phospholipase (PL) A2 (1 nmol PACOCF3), PLC (30 pmol U73122), PKC (1 nmol GF109203X), p38 mitogen-activated protein kinase (MAPK; 30 nmol SB203580), extracellular signal-regulated kinase (ERK1/2; 30 nmol PD98059), c-Jun N-terminal kinase (JNK; 30 nmol SP600125), or vehicle, to assess their influence on the hyperalgesic response. The mechanical hyperalgesia caused by ET-1 started 2 hrs after injection, peaked at 5 hrs (PBS, 29 ± 0.5 g; ET-1, 17 ± 1.3 g) and lasted up to 8 hrs. The inhibitors of PLC, PKC, p38 MAPK, ERK1/2, and JNK caused long-lasting reductions of the mechanical hyperalgesia (inhibitions at 4 hrs of 100%, 90%, 97%, 90%, and 100%, respectively), but the PLA2 inhibitor reduced hyperalgesia only at 4 hrs (by 58%). Thus, mechanical hyperalgesia triggered by ET-1 in the rat hind paw depends importantly on signaling pathways involving PLC, PKC, p38 MAPK, ERK1/2, and JNK, whereas the contribution of PLA2 is relatively minor.
Introduction
Endothelin (ET)-1 and other peptides of the ET family are produced by many cell types, including central and peripheral neurons, and cause a broad spectrum of potent effects via activation of specific G-protein–coupled ETA and ETB receptors (1, 2). Depending on cell type, both ET receptor subtypes can be coupled to multiple intracellular signaling mechanisms, including those involving phospholipases (PL) A2, PLC, and PLD; adenylyl and guanylyl cyclases, and mitogen-activated protein kinases (MAPK; Refs. 3–5).
Studies show that ET-1 causes spontaneous pain (or overt nociception) and hyperalgesia (i.e., enhanced responsiveness to noxious stimuli) in animals and humans. Thus, volunteers report burning pain and hyperalgesia to tactile stimuli when ET-1 is injected intradermally into the forearm (6). In rodents, this peptide evokes abdominal writhing (via ETA and ETB receptors) when given intraperitoneally, but causes overt nociception as well as hyperalgesia to noxious chemical and thermal stimuli via ETA receptors if injected into the hind paw (6–10). ET-1, which is expressed in small-to medium-diameter neurons and satellite cells in dorsal root ganglia (11, 12), can also activate ETA receptors on sensory C-fibers of the sciatic nerve to cause paw flinches (13) or in the knee joint to cause articular incapacitation (14) in rats. On the other hand, ET-1 injection into the plantar surface of the rat hind paw has been reported to elicit mechanical hyperalgesia exclusively via ETB receptors (15). However, we have seen 1 that this effect of ET-1 in rats can be reduced by local injection of either ETA or ETB receptor antagonists, as occurs in mice (10).
Motta EM, Rae GA. Unpublished observations.
Few studies have detailed the intracellular and trans-cellular signaling mechanisms underlying the alterations in nociceptive responsiveness triggered by ET-1. In this regard, the persistent mechanical hyperalgesia induced by ET-1 in the rat hind paw is potentiated by rolipram (a phosphodiesterase blocker) and blocked by staurosporine or calphostin C (both PKC inhibitors), but not by indomethacin (a cytochrome c oxidase [COX] inhibitor), dexamethasone (a glucocorticoid), atenolol (a β-adrenoceptor antagonist), or H89 (a PKA inhibitor; Ref. 15). The current study assesses the putative contributions of PLA2, PLC, PKC, and several MAPKs, including the extracellular signal-regulated kinases (ERK)-1 and ERK2, p38 MAPK, and c-Jun N-terminal kinase (JNK), to ET-1–induced mechanical hyperalgesia in rats.
Materials and Methods
Animals.
Experiments were conducted on male Wistar rats weighing 200–240 g, housed five to a cage at 22 ± 2°C on a 12:12-hr light:dark cycle (lights on at 0700 hrs), with free access to laboratory chow and tap water. Experimental procedures were approved beforehand by the institution’s Ethics Committee on Experimental Use of Animals.
Drugs and Solutions.
The following drugs were used: ET-1 (American Peptide Co., Sunnyvale, CA), GF109203X, U73122, PACOCF3, SB203580, PD98059, and SP600125 (all from Tocris, Ellisville, MO). Stock solutions of ET-1 were prepared in phosphate-buffered saline (PBS; Sigma Chemical Co., Saint Louis, MO), those of other drugs in absolute ethanol or dimethylsulfoxide (which, injected at maximal final concentrations of 0.5% and 0.1%, respectively, caused no effects per se). All stock solutions were stored at −20°C and diluted to the desired concentration in PBS just before use.
Mechanical Hyperalgesia.
The threshold for nociceptive responsiveness to mechanical stimuli applied to the hind paw was assessed using an electronic version of the Von Frey test (dynamic plantar aesthesiometer, model 37400; Ugo Basile, Milan, Italy). Each rat was placed in a Plexiglas chamber (28 × 40 × 35-cm, wire mesh floor). Fifteen minutes later, a servo-controlled mechanical stimulus (a pointed metallic filament) was applied to the plantar surface repeatedly, at 5-min intervals, which exerted a progressively increasing punctate pressure, reaching up to 30 g within 20 s. The pressure evoking a clear voluntary hind-paw withdrawal response (usually close to 30 g) was recorded automatically and taken as the mechanical nociceptive threshold index. Mechanical threshold was always assessed three times at each time point to yield a mean value. Rats were habituated to the full procedure for 30 min/d on two consecutive days and experiments were conducted on the third day only with those rats that effectively responded to mechanical stimulation (cut-off time 40 secs).
Drug Treatments.
After determining the basal mechanical thresholds, each animal received an intraplantar (ipl) injection of selective inhibitors of either PLA2 (1 nmol PACOCF3), PLC (1 or 30 pmol U73122), PKC (1 nmol GF109203X), p38 MAPK (30 nmol SB203580), ERK1/2 (30 nmol PD98059), or JNK (30 nmol SP600125), or 30 μl of the corresponding vehicle. A second ipsilateral ipl injection of either 30 pmol ET-1 or 20 μl PBS was applied 15 mins later, and the mechanical threshold for nociceptive responsiveness was evaluated repeatedly at 1-hr intervals for up to 8 hrs and, in some cases, once again at 24 hrs. In all experiments, responses of drug-treated animals were always assessed in parallel with responses of vehicle-treated (i.e., day-matched control) rats, to minimize the interference of possible fluctuations in responsiveness. Doses were selected on the basis of previous studies (16, 17) and preliminary experiments carried out in the laboratory.
Statistical Analysis.
All results are presented as mean ± SEM of at least five animals. The data were analyzed using two-way analysis of variance (ANOVA; treatment and time as factors) and, if significant interactions were found, post hoc comparisons were made using Student-Newman-Keuls t test. Significance was set at P < 0.05. Because the results of the various vehicle-pretreated PBS-treated control groups did not differ statistically from each other, these data were pooled (total N =30). The same applies for the different vehicle-pretreated ET-1–treated groups (total N = 35).
Results
Ipl injection of 30 pmol ET-1 induced a slowly developing and prolonged unilateral reduction in the mechanical threshold for nociceptive responsiveness of the injected paw. The response started 2 hrs after injection, plateaued between 3 and 6 hrs, subsided partially up to 8 hrs (Fig. 1A), and was absent 24 hrs after treatment (data not shown). Moreover, this response to ET-1 was largely unaffected by previous ipsilateral ipl injection of 1 nmol of the PLA2 inhibitor, PACOCF3, which transiently attenuated hyperalgesia at 4 hrs only (Fig. 1A). In contrast, similar pretreatment with 30 pmol of the PLC inhibitor, U73122, abrogated the ET-1–induced mechanical hyperalgesia for up to 5 hrs, but its effect subsided thereafter (Fig. 1B). Marked inhibition of hyperalgesia was also seen with 1 nmol of the PKC inhibitor, GF109203X, but the onset of its effects were somewhat delayed (3 hrs) in comparison with U73122 (Fig. 1C). On the other hand, previous local inhibition of p38 MAPK with 30 nmol SB203580, of ERK1/2 with 30 nmol PD98059, or of JNK with 30 nmol SP600125 each reduced ET-1–induced mechanical hyperalgesia substantially (Fig. 2A, B, and C, respectively). The net peak inhibitions afforded by pretreatment with SB203580, PD98059, and SP600125 reached 97%, 95%, and 100%, respectively. None of the pretreatments administered caused any gross alterations in consciousness, locomotor activity, or behavior during the full length of the experiments.
Discussion
The current results fully confirm that ipl-administered ET-1 causes persistent unilateral mechanical hyperalgesia when injected into the hind paw of rats (15), as it also does in mice (10). More importantly, the results furnish new insight into some of the possible signaling pathways implicated in this phenomenon.
Although ET receptors can be coupled to PLA2 in several cell types (18), the mechanical hyperalgesia in response to local ET-1 injection was only transiently reduced by PLA2 inhibition via PACOCF3. This finding suggests that this local effect of ET-1 is dependent only to a limited extent on arachidonic acid mobilization for conversion to hyperalgesic prostaglandins and/or leukotrienes. Indeed, this view is strengthened by a report that the ETB receptor–mediated mechanical hyperalgesia caused by ET-1 in rats is fully resistant to blockade by the COX inhibitor, indomethacin, or dexamethasone (15), although glucocorticoids act on many additional targets in addition to PLA2 and COX.
Conversely, the PLC-PKC pathway, another frequently encountered signaling mechanism coupled to ET receptor activation (19), seems to play a major role in mechanical hyperalgesia triggered by ET-1. Substantiating this view, we observed that the highly selective PLC inhibitor, U73122, completely suppressed the development of the response to ET-1 for up to 5 hrs. Furthermore, blocking this pathway further downstream with the PKC inhibitor, GF109203X, resulted in a similar but somewhat delayed effect, confirming previous findings that the PKC inhibitors staurosporine and calphostin C are antihyperalgesic against ET-1 in a different model of mechanical stimulation (15). Activation of the PLC-PKC pathway can lead to phosphorylation of several targets, including ion channels and transporters, matrix metalloproteinases, and activation of small G-proteins of the Ras family (20).
Small G-proteins of the Ras family can recruit distinct MAPK pathways, including p38 MAPK, ERK1/2, and JNK, which target various transcription factors controlling protein expression, in addition to causing nontranscriptional short-term changes in cell function (21). These MAPK pathways are important regulators of nociceptive sensitivity in various models of inflammatory pain and hyperalgesia to mechanical and thermal stimuli (22). The current study reveals, to our knowledge, for the first time, that potent and selective inhibitors of p38 MAPK, ERK1/2, and JNK can each markedly reduce ET-1–induced mechanical hyperalgesia. Therefore, small G-proteins of the Ras family play a pivotal role in coupling ET receptors to enhancement of mechanical nociceptive sensitivity. The putative role for PKB (Akt) proteins, activated via phoshoinositide 3-kinase signaling and transactivation of neurotrophin tyrosine kinase receptors (21), in the PKC-dependent hyperalgesic response to ET-1 remains to be determined.
Hind-paw mechanical hyperalgesia induced by ET-1 in the rat has been attributed exclusively to stimulation of local ETB receptors (15). Indeed, we found that similar responses are elicited by 3 pmol of the selective ETB receptor agonist, sarafotoxin (S6c; data not shown). However, we also observed that local injection of selective ETA or ETB receptor antagonists (10 nmol BQ-123 and 10 nmol BQ-788, respectively) can each partially and transiently reverse established mechanical hyperalgesia promoted by ET-1 2 . This discrepancy might reflect differences in the characteristics of mechanical stimulation used in both studies, that is, plantar stimulation with progressively increasing pressure (present study) versus application of a constant pressure (15).
Motta EM, Rae GA. Unpublished observations.
It seems relevant to mention that, in addition to its pronociceptive functions, ET-1 can also display antinociceptive actions. Thus, the ETA receptor–mediated overt pain caused by ET-1 administration in the rat hind paw can be limited by activation of ETB receptors on keratinocytes coupled with release of antihyperalgesic/analgesic β-endorphin (23). Moreover, ET-1 causes analgesia when microinjected into the lateral cerebral ventricle of mice, and selective knockout of the END1 gene encoding ET-1 in neurons sensitizes this species to noxious stimuli (24, 25). In the present study, we chose to administer the various blockers of signaling pathways into the hind paw. Therefore, we speculate that these various agents acted locally to interfere with nociceptive signal processing, rather than via systemic or centrally mediated actions. Nonetheless, more studies are clearly needed to define the precise location and identity of the receptors responsible for this hyperalgesic action of ET-1 in the rat hind paw.
In conclusion, the current study provides evidence that mechanical hyperalgesia triggered by ET-1 in the rat hind paw depends importantly on signaling pathways involving PLC, PKC, p38 MAPK, ERK1/2, and JNK, whereas the contribution of PLA2 is relatively minor. It remains to be seen whether these mechanisms are differentially coupled to ETA and ETB receptors and also contribute to hyperalgesia in other forms of noxious stimuli. Finally, considering that the ET system can play roles in pain associated with immune, inflammatory, neoplasic, and neuropathic states (8, 26–28), these results might be useful to identify potential targets for better management of pain.
Influence of inhibitors of PLA2 (PACOCF3; A), PLC (U73122; B), or PKC
(GF109203X; C) on mechanical hyperalgesia induced by ET-1 in the rat hind paw. The
inhibitors or corresponding vehicles were injected ipl, at the doses indicated, 15
mins before ipsilateral ipl injection of ET-1 or PBS. The threshold for mechanical
noxious stimulation was evaluated before treatment (time zero) and then every hour
until 8 hrs after ET-1 (or PBS) administration. Values represent the mean ± SEM of at
least five observations. Asterisks and fences indicate P < 0.05
relative to corresponding values of vehicle + PBS or vehicle + ET-1 groups,
respectively (two-way ANOVA followed by Student-Newman-Keuls t
test). Influence of inhibitors of p38 MAPK (SB203580; A), ERK1/2 (PD98059; B), or JNK
(SP600125; C) on mechanical hyperalgesia induced by ET-1 in the rat hind paw. The
inhibitors or corresponding vehicles were injected ipl, at the doses indicated, 15
mins before ipsilateral ipl injection of ET-1 or PBS. The threshold for mechanical
noxious stimulation was evaluated before treatment (time zero) and then every hour
until 8 hrs after ET-1 (or PBS) administration. Values represent the mean ± SEM of at
least five observations. Asterisks and fences indicate P < 0.05
relative to corresponding values of vehicle + PBS or vehicle + ET-1 groups,
respectively (two-way ANOVA followed by Student-Newman-Keuls t
test).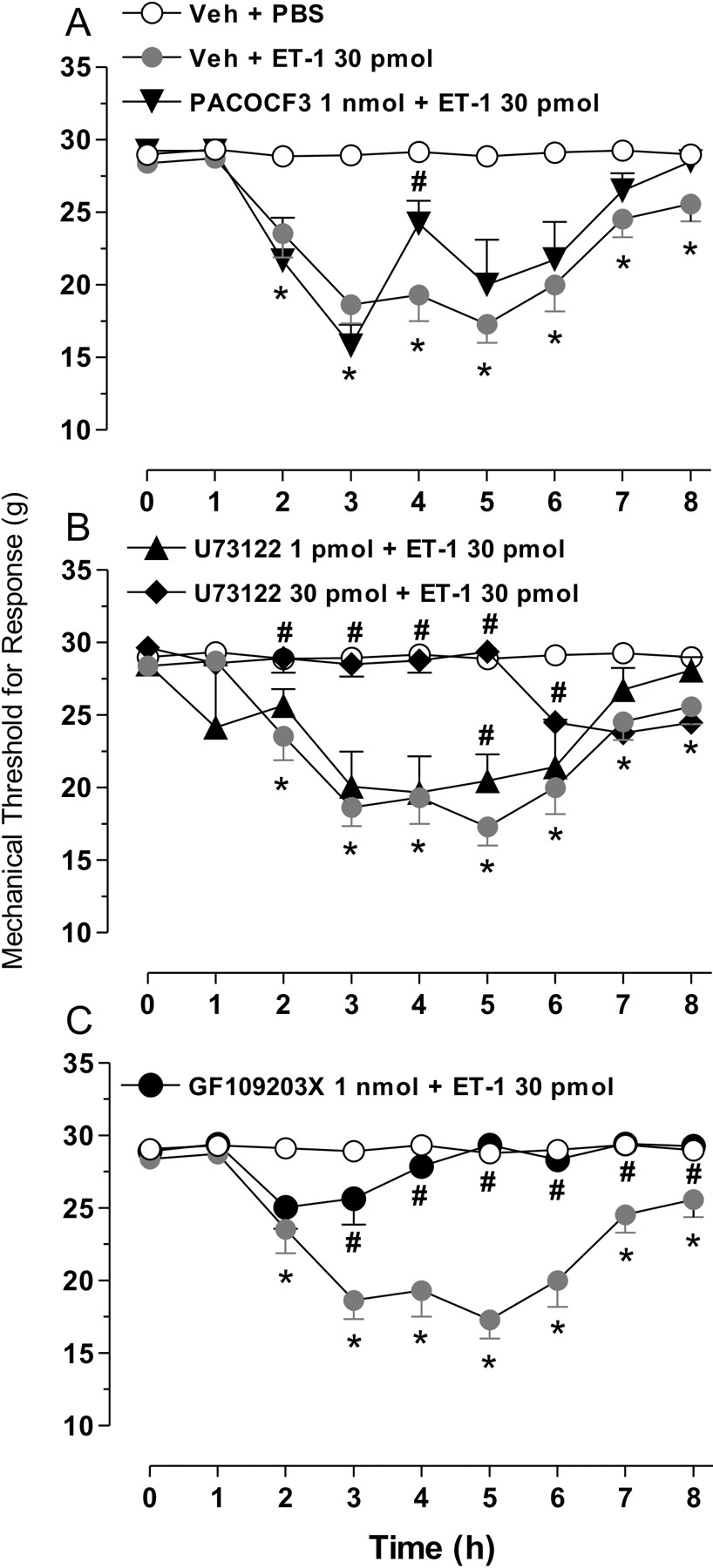

Footnotes
The study was supported by the Brazilian National Research Council (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Fundação de Amparo à Ciência e Tecnologia do Estado de Santa Catarina (FAPESC), and the Pronex program of the Brazilian Ministry of Science and Technology. E.M.M. is the recipient of a CNPq doctoral scholarship.