
Abstract
Cerebral malaria (CM) remains a deadly complication of Plasmodium falciparum infection, and children are at high risk of developing encephalopathy as a result of CM. This is probably a consequence of the activation of many of the inflammatory cytokines as well as the glial cells and the vascular endothelium in the brain. We have previously demonstrated that there is a striking reduction in cerebral blood flow by magnetic resonance imaging when mice are infected with Plasmodium berghei ANKA (PbA), and we now demonstrate a possible role for endothelin (ET-1) in the pathogenesis of CM. The brains of female C57BL/6 mice with PbA infection were examined at Day 5 for the expression of ET-1, endothelin converting enzyme (ECE), and the endothelin receptors A and B (ETA and ETB) by both reverse transcription–polymerase chain reaction (RT-PCR) and quantitative real-time PCR. ET-1 and ECE mRNA expression was markedly increased by RT-PCR in PbA-infected mice. Real-time quantitative PCR demonstrated a 3-fold increase in ET-1 (P < 0.05) and a significant increase in ETA and ETB expression (P < 0.05) in PbA-infected mice. Histopathology bof PbA-infected mice demonstrated a transformation in the morphology of microglial cells and clustering of these cells consistent with activation. Though the full impact of ET-1 on CM remains to be elucidated, these findings demonstrate that in the murine model, there is a significant increase in ET-1 and its components, which is associated with the vasculopathy and immunopathology of CM.
Introduction
Malaria is a very important disease entity in the developing world and remains a fatal disease in over a million children in sub-Saharan Africa (1). Cerebral malaria (CM) in humans is the most life-threatening complication of infection with Plasmodium falciparum and usually results in an encephalitic picture with ataxia, seizures, inability to localize painful stimuli, hemiplegia, and, eventually, coma and death (1, 2). Despite extensive studies on the pathogenesis of CM, there are significant gaps in our understanding of the pathogenesis of this disease. This is partially due to the fact that the pathologic process is difficult to observe in humans, in whom investigations are essentially limited to postmortem samples (2). We have previously demonstrated by magnetic resonance imaging (MRI) that there is a striking reduction in cerebral blood flow when mice are infected with Plasmodium berghei ANKA (PbA) (3). Such human and animal studies have indicated that encephalitis due to CM is mostly an immunopathologic process due to the production of cytokines, resulting in the activation of the cerebral vascular endothelium. This activation results in the sequestration of erythrocytes and mononuclear leukocytes and platelet adhesion (2, 4) as well as in the activation of an endogenous central nervous system immune response with astrogliosis and glial cell activation (5).
Animal studies have demonstrated that there is extensive damage in the brains of infected subjects, including ischemia, hemorrhage, breakdown of the blood-brain barrier, and swelling (6). It has long been proposed that these changes occur as a result of the host inflammatory response to infection as well as blockage of cerebral vessels by the sequestered inflammatory cells and infected red blood cells (RBCs) and an alteration in metabolic markers in the brain (1, 3, 4). We and others (3, 7) have now demonstrated that in mice infected with CM, there is a marked decrease in cerebral blood flow at advanced stages of the disease, as measured by MRI, and that this decrease is directly correlated with a significant decrease in N-acetyl aspartate levels in areas of the brain, indicative of neuronal damage.
Vasculopathy appears to be an important component of CM. Such vasculopathies are associated with endothelial cell damage, activation of vascular cell adhesion molecules, and an associated breakdown in the blood-brain barrier (8). To investigate the vasculopathy of CM, we studied the role of endothelin (ET-1) in the pathogenesis of this disease.
ET-1 is a 21–amino acid peptide. It is a very potent vasoconstrictor with mitogenic properties (9). It is synthesized by vascular endothelial cells as well as by a variety of other cells, including neurons and astrocytes in the brain at low basal levels (9, 10); however, the level of expression of ET-1 in tissues is upregulated in the presence of certain processes and with stress to the endothelium (9). In addition to its effects on the vascular smooth muscle cells, resulting in a reduction of blood flow, ET-1 is also capable of modifying the blood-brain barrier, as shown by a decrease in cerebral edema when the receptor is antagonized (11). We hypothesized that a consequence of the damage to the endothelium by the malarial parasite is that the expression of ET-1 would be upregulated and that the increased levels of ET-1 would result in the decrease in cerebral blood flow seen in CM.
Materials and Methods
Infection of Mice.
Six- to eight-week-old female C57BL/6 mice (Taconic, Germantown, NY) were infected with 105 PbA-parasitized RBCs via intraperitoneal injection. Infected mice were compared to age- and sex-matched uninfected controls (N = 6 for each group). The parasitized blood was obtained from previously infected mice on the day of infection and was diluted in phosphate-buffered saline to provide 105 PbA parasitized RBCs in an 0.2-ml injection. At Day 5 postinfection, the mice were euthanized using carbon dioxide and the brains were harvested, frozen in liquid nitrogen, and then stored at −80°C for future analysis. For histopathology, brains were immediately placed in buffered formalin and stored at room temperature.
RNA Extraction and cDNA Determination by Reverse Transcription–Polymerase Chain Reaction (RT-PCR) and Real-Time Quantitative PCR.
Using the TRIzol reagent, RNA was extracted from brain homogenates according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA). Five nanograms of RNA was used to prepare cDNA: we used a Superscript II reverse transcriptase kit (Invitrogen), per the manufacturer’s manual, employing a 50-min incubation at 42°C in a RT mixture consisting of 0.5 mM deoxynucleoside triphosphates; 20 mM dithiothreitol; 30 mM Tris-HCl, pH 8.3; 75 mM KCl; 3 mM MgCl2; 500 ng oligo dT, and 200 U of superscript RT RNase H-reverse transcriptase.
Quantitative PCR was performed using primers for ET-1 (ET-1 forward: 5′CTGCCACCTGGACATCATC 3′; ET-1 reverse: 5′TCCTTCCTTCCACCAGCTG 3′, which amplified a 280–base pair [bp] endothelin-1 gene fragment); ETA (ETA forward: 5′CCCTTAGTGAGCACCTCAAA 3′; ETA reverse: 5′CACACCGGTTCTTATCCATC 3′, which amplified a 147-bp endothelin type A receptor gene fragment); and ETB (ETB forward: 5′CGCTCTGTATTT-GGTGAGCA 3′; ETB reverse: 5′AGTGAGATTCGGC-GAGTGTT 3′). The quantitative PCR was run using 2 mM MgCl2 and the PCR Sybr Green Master Mix (Roche Applied Science, Indianapolis, IN) in a final volume of 20 μl. Reaction mixes were loaded into Roche Light Cycler Capillaries, capped, centrifuged for 10 secs at 2000 rpm, and placed in the Light Cycler (Roche). The reaction was run at 95°C at 20°C/sec ramp for 10 mins, followed by 45 cycles of 95°C at 20°C/sec ramp for 5 secs; 58°C at 20°C/sec ramp for 7 secs; and 72°C at 20°C/sec ramp for 20-sec hold. The fluorescence intensity was acquired at 72°C. The third phase of the real-time PCR was the melt phase, which was performed at 95°C at 20°C/sec ramp for 0-sec hold, then 65°C at 20°C/sec ramp for 30-sec hold, and finally 95°C at 0.1°C/sec ramp for 0-sec hold. During the melting phase, the acquisition setting was set at “continuous.” Finally, a cooling phase of 30 secs at 40°C at 20°C/sec was used. Data were acquired and analyzed with Light Cycler (version 3.0) software. For each run, there was a negative control with no added DNA template.
To generate PCR standards, genomic DNA was isolated from control murine brain tissues using the Qiagen DNeasy kit following the manufacturer’s protocol (Qiagen, Inc., Valencia, CA). A standard curve in the range of 5 pg to 50 ng for the quantification of respective products by quantitative PCR was developed using above-described primers and conditions. The result was normalized by dividing the number of copies of mRNA (cyclins) by the number of copies of GAPDH mRNA for each sample. The primer sequence used for GAPDH forward was 5′AACTT TGGCATTGTGGAAGG 3′ and for GAPDH reverse was 5′ACACATTGGGGGTAGGAACA 3′, amplified on a 225-bp GAPDH gene fragment. The conditions for quantitative PCR used for quantification of GAPDH are the same as above. A t test was used for statistical analysis.
RT-PCR was performed using Taq polymerase (Gibco-BRL, Gaithersburg, MD) in a PTC-100 thermal cycler (MJ Research, Watertown, MA). The reaction conditions comprised 35 cycles of 1 min at 94°C, 1 min at 54°C, and 2 mins at 72°C, with a final extension step of 7 mins at 72°C. The primers used for the quantitative PCR of ET-1 were also used for this reaction. For ECE, the primers used were as follows: ECE forward: 5′ATGACGCCGCCCATGGTGAAC 3 ′, and ECE reverse: 5′TGGTTGGGCTAAGACATAAC 3′. For each set of primers, a negative sample (water) was run in parallel. The PCR products were subsequently separated by acrylamide gel electrophoresis and stained with silver nitrate.
Immunohistochemistry.
Brain tissue was fixed in buffered formalin, embedded in paraffin, and sectioned into 4-μm sections. Sections were then deparaffinized and boiled at 95°C for 20 mins in sodium citrate solution (DAKO, Carpinteria, CA) for antigen retrieval. Immunohistochemistry was performed using rabbit antibody to ionized calcium binding adaptor molecule 1 (Iba1) (Wako Chemicals USA, Inc., Richmond, VA) at a 1:300 dilution, incubated overnight at 4°C, as well as mouse immunoglobulin G1 against unphosphorylated neurofilament epitope SMI32 (Sternberger Monoclonals, Baltimore, MD) at 1:500 dilution, with overnight incubation at 4°C. A standard ABC method was used for the secondary antibodies (anti-mouse and anti-rabbit) using a kit from Vector Immunolab (Burlingame, CA) at 1:200 with 1-hr incubation. Slides were counterstained with hematoxylin after immunolabeling.
MRI.
MRI of the mice was performed using a 9.4T horizontal bore Varian Inova system at 6 days postinfection for both control (n = 4) and PbA-infected (n = 4) mice. The blood flow values found in uninfected mice were comparable to those obtained from a database of uninfected C57BL/6 mice using a similar protocol (n = 8), where the average value of cerebral blood flow (CBF) was 94 ± 26 ml/100 g/min (12). CBF measurements were acquired using a flow-alternating arterial inversion spin-labeling method. Single voxel proton spectroscopy (18 μl) was used to measure the relative concentrations of N-acetyl aspartate (NAA), creatine (Cr), and choline from the caudate and putamen. Student’s t test was used for statistical analysis, as previously described (12).
Results
Survival and Parasitemia.
As demonstrated in Figure 1A, murine CM is a fatal disease, with mortality occurring between 7 and 10 days. At the time of death, parasitemia usually measures from 10%–20% (Fig. 1B). This phenomenon was observed in several mice during interrodent passage of the parasitized blood and is well cited in the literature (2, 5, 6). There was no parasitism or mortality observed in the uninfected mice.
MRI.
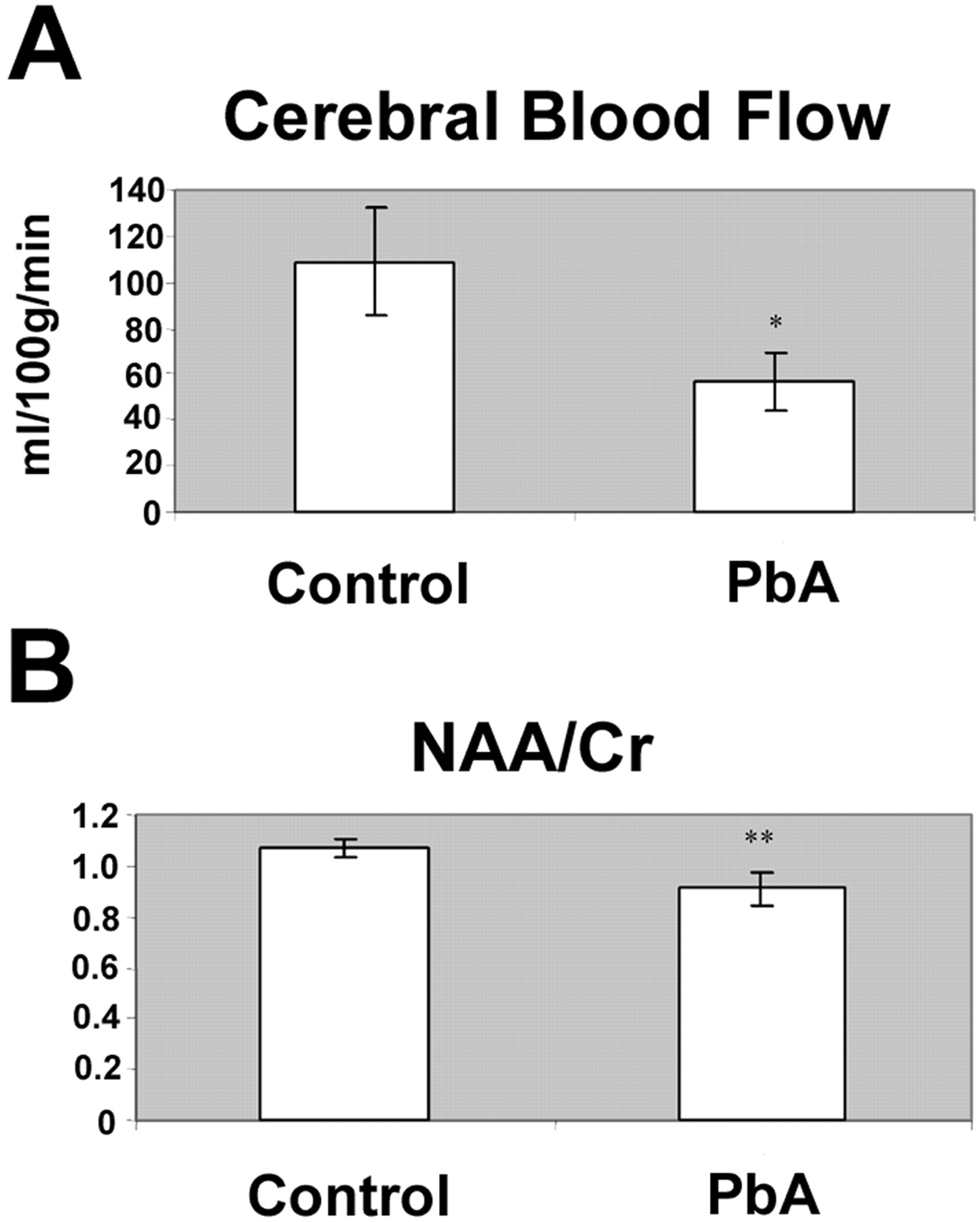
Consistent with our previous data, there is a significant reduction in CBF in mice at advanced stages of disease from CM, when compared to controls (3). The average CBF in control mice was 109 ± 23 ml/100 g/min, versus 57 ± 13 ml/100 g/min in infected mice, which corresponds to a 48% decrease in CBF associated with CM (P < 0.05) (Fig. 2A). Similarly, we observed a 15% reduction in the NAA to Cr ratio in the caudate putamen of the infected mice; 0.91 ± 0.07 as opposed to 1.07 ± 0.04 in control mice (P < 0.01) (Fig. 2B). We found that there was in fact a direct correlation between the CBF and NAA to Cr ratio (data not shown) (3), which may indicate that a decrease in blood flow to the brain is associated with the neuronal damage that occurs with CM.
Endothelin Studies.
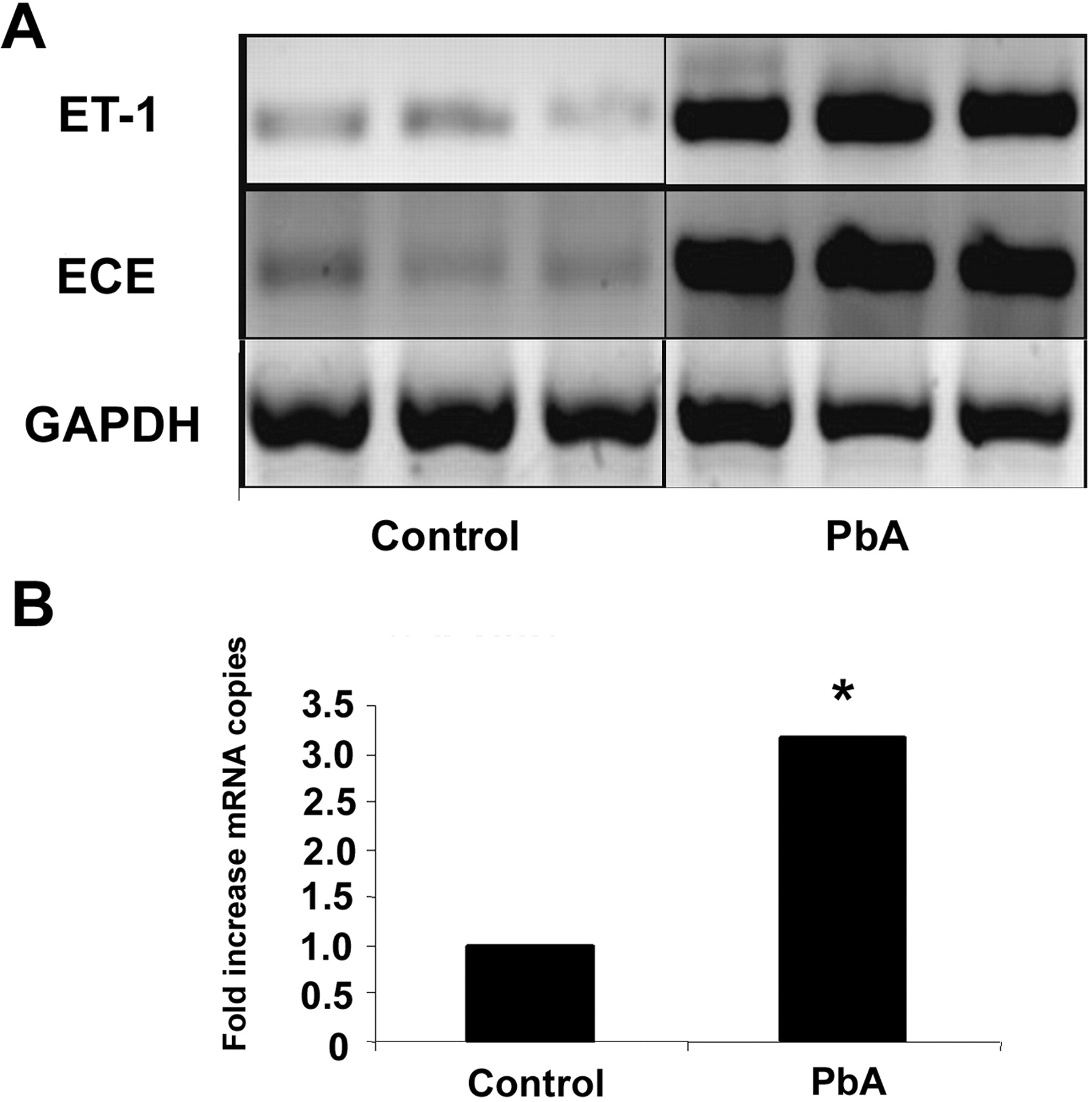
We observed a significant increase in several components of the endothelin pathway in these mice. Both ET-1 and ECE mRNA expression were markedly increased by RT-PCR (Fig. 3A), and when analyzed quantitatively by quantitative PCR, we observed a 3-fold increase in the mRNA expression of ET-1 in infected mice compared to uninfected controls (P < 0.05; Fig. 3B). Correspondingly, when we looked at the endothelin receptors by quantitative PCR, we were also able to demonstrate a 1.5-fold increase in the mRNA expression of ETA in the infected mice (P < 0.05; Fig. 4A) as well as a 6.4-fold increase in the mRNA expression of ETB in these mice (P < 0.05; Fig. 4B).
Immunohistochemistry.
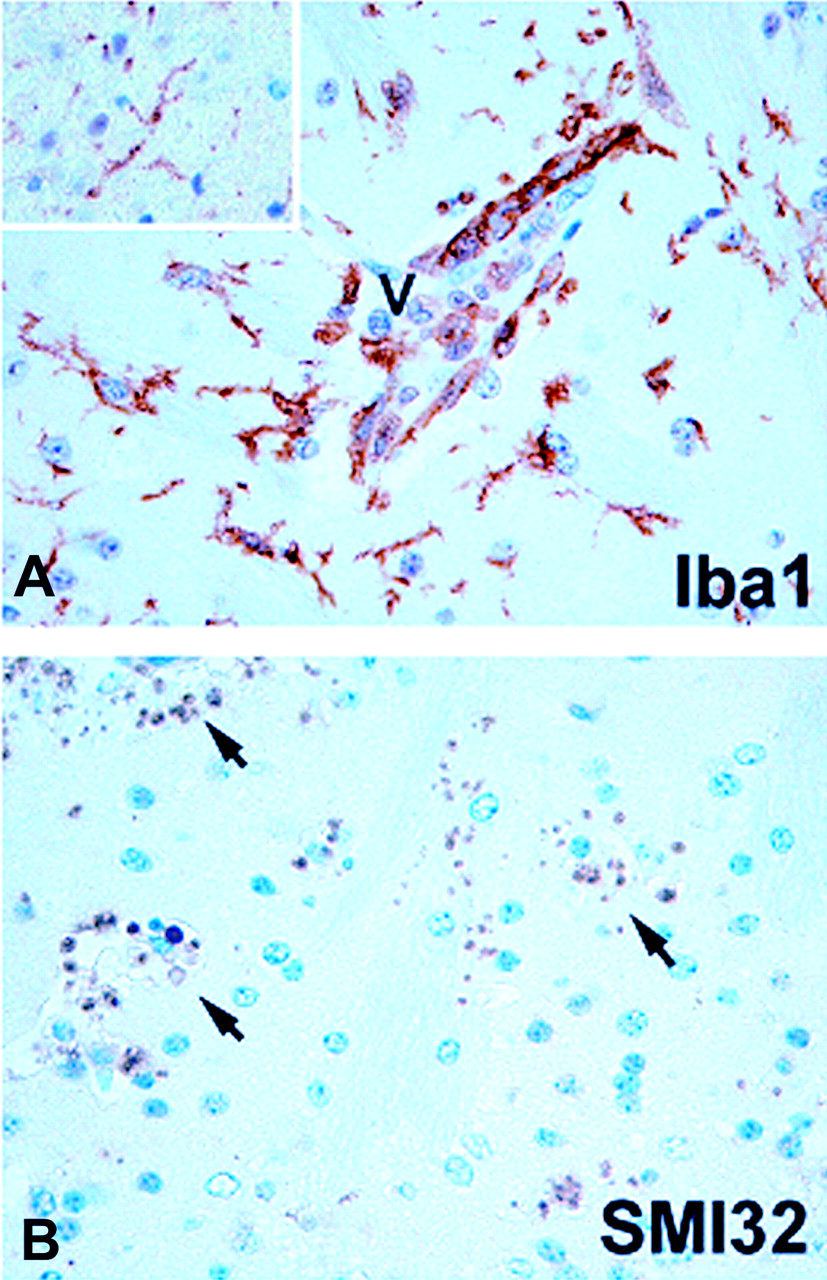
In the areas of cerebral hemorrhage, there was marked damage of the neuronal axons in the infected mice, demonstrated by immunohistochemistry as clusters of SMI32+ profiles varying in size and reaching the size of an erythrocyte (Fig. 5B). The observed profiles are consistent with transected axon terminals that have abnormal expression of unphosphorylated neurofilament epitope (13). Moreover, there was considerable evidence of microglial activation in the areas of damage, with hypertrophy as well as retraction and thickening of the processes in addition to formation of clusters (Fig. 5B). In contrast, microglial cells in uninfected brains were highly ramified and had delicate morphology (Fig. 5A, insert).
Discussion
CM is an important cause of morbidity and mortality, especially among children in sub-Saharan Africa (1). The precise etiology is not clear; however, there are suggestions (2–4, 6, 7) that alterations in the integrity of the blood-brain barrier and vascular dysfunction may contribute to the pathogenesis of CM. Although the murine model of CM does not totally recapitulate the entity in humans, it has nevertheless become an important model to study the pathogenesis of CM (1–8).
In the present investigations, CBF was reduced by almost 50% in ANKA strain–infected mice. This was accompanied by a corresponding 15% reduction in the ratio of NAA to Cr. NAA has been widely used as a marker of neuronal loss and injury in a variety of pathologies, including epilepsy (14), Alzheimer’s disease (15), stroke (16), and multiple sclerosis (17). NAA is synthesized only in neuronal mitochondria (18), correlated with bioenergetic status (19), and in epilepsy its decrements can be reversed with return of function (20). Thus, a decrease in NAA levels usually reflects a mixture of both neuronal loss and recent or ongoing neuronal injury/dysfunction, as reflected by its correlation with GFAP expression and gliosis (21). In addition, we found abnormalities and damage in the neuron/axon compartment. Immunohistochemical studies demonstrated numerous areas containing SMI32+ axon profiles in the brains of infected animals. Neurofilaments of healthy axons are heavily phosphorylated under normal circumstances and do not stain with SMI32 antibodies. Thus, SMI32 immunoreactivity provides a sensitive marker for axonal pathology (13).
Previous MRI data demonstrated that focal intrastriatal injection of tumor necrosis factor–α (TNF-α) leads to significant decreases in blood volume (22). The observed CBF reduction in the current study would thus be consistent with cytokine-induced vasoconstriction. In this regard, it has been previously demonstrated that increased expression of TNF-α in the brain, such as that which occurs during CM, induces a significant reduction in CBF, one that is endothelin dependent (22). Moreover, TNF-α facilitates the release of ET-1 by endothelial and epithelial cells (23).
In the present study we demonstrated that there was a significant increase in the various components of the endothelin pathway that were observed concomitantly with a marked reduction in CBF and neuronal cell damage. The increased ET-1 levels in the brain correlated with histologic evidence of glial cell activation. These findings are consistent with previous examination of mouse retinal tissue in CM, which demonstrated alterations in the morphology of microglial cells upon activation (5).
ET-1 in the brain is synthesized by a variety of cells, including neurons and glial cells in addition to the vascular endothelial cells, and it is widely distributed in the brain (9, 10); thus, it is not surprising that the levels of ET-1 would be elevated with the activation and damage of these cells. The exact contribution of each of these cell types in the synthesis of ET-1 during CM remains to be elucidated. The increase in ET-1 in the brain of mice with CM correlates well with evidence of increased plasma levels of big ET-1 in patients with acute complicated P. falciparum disease (23). ET-1 is thought to be involved in a variety of pathologic processes that occur in CM. In this regard, ET-1 is a potent vasoconstrictor that may also contribute to increased platelet aggregation. Moreover, ET-1 increases the expression of leukocyte adhesion molecules and the synthesis of inflammatory mediators, which may contribute to vascular dysfunction and to loss of integrity of the blood-brain barrier (11, 24).
Overall these data indicate that the increase in ET-1 that accompanies murine CM may be a contributing factor in the reduction of CBF and the ensuing neuronal damage observed. Further investigation of the ET-1 pathway in experimental models and humans is warranted, as the endothelin pathway may provide important and unique targets for adjunctive therapy in CM, limiting the neurologic damage associated with malaria infections.
(A) Parasitemia in Plasmodium berghei ANKA infection. At the time of
death, the parasitemia is usually in the midteens. (A) Cerebral blood flow is markedly decreased in mice infected with
Plasmodium berghei ANKA (PbA) when compared to uninfected control
mice. (B) The reduction in blood flow corresponds with the reduction in the caudate
putamen of NAA:Cr ratio at Day 6 postinfection. *, P < 0.05;
(A) The components of the endothelin system are elevated in whole-brain lysates of
mice infected with Plasmodium berghei ANKA (PbA). (Panel A) RT-PCR
demonstrates a marked increase in the mRNA expression of endothelin (ET-1) and
endothelin-converting enzyme (ECE) in infected mice when compared to control mice at
Day 5 postinfection. (B) Quantitative PCR of ET-1 demonstrates a 3-fold increase in
mRNA expression of ET-1 in infected mice (N = 6) compared to controls
(N = 6). *, P < 0.05. (A) Quantitative PCR of the endothelin receptor A (ETA) demonstrates a
1.5-fold increase in the expression of the receptor (N = 6 in both
groups). (B) Likewise, quantitative PCR demonstrates a 6.4-fold increase in the mRNA
expression of the endothelin receptor B (ETB) (N = 5 for
infected and N = 6 for controls). *, P <
0.05. (A) Iba1 immunohistochemistry demonstrates ramified microglial cells in control brain
(insert). In murine cerebral malaria (CM) due to Plasmodium berghei
ANKA infection, microglial cells are transformed to a less-ramified, macrophage-like
shape in areas of axonal damage. (B) Immunohistochemistry for unphosphorylated
neurofilament epitope (SMI32) demonstrates clusters of SMI32+ profiles,
consistent with damaged axons (arrows). In both control and CM mice, SMI32 stains
neurons and dendrites in the gray matter (normal staining not shown). Color figure
available in on-line version of the journal.

Footnotes
This work was supported in part by the Dana Foundation under the Dana Program in Brain and Immuno-imaging (H.B.T.) and by the following National Institutes of Health (NIH) grants: AI-12770, AI-052739, HL-073732 (H.B.T.), EB-001787 (H.P.H.), DK-55758, and CA-112023 (P.E.S.). S.C.L. was supported by the Albert Einstein College of Medicine CFAR P30 AI051519. M.S.D. was the recipient of the IDSA ERF/NFID Colin L. Powell Minority Postdoctoral Fellowship in Tropical Disease Research sponsored by GlaxoSmithKline, and she was supported by the NIH Training Grant in Mechanisms of Cardiovascular Diseases (T32 HL-07675).
1
Contributed equally to this work.
Acknowledgements
We would like to thank Dr. Meng-Liang Zhao for the immunohistochemistry and Drs. Naoyuki Miyasaka and Kan Takahashi for assistance with the MRI.