
Abstract
This study was designed to determine the effect of selenium (Se) deficiency on the immune response to infection with a virulent strain of influenza virus, influenza A/Puerto Rico/8/34. Previous work in our laboratory demonstrated that Se-deficient mice infected with a mild strain of influenza virus, influenza A/ Bangkok/1/79, developed much more severe lung pathology compared with Se-adequate mice. Immune function was altered in the Se-deficient mice, and the viral genome changed to a more virulent genotype. In this study, we tested whether Se deficiency would have a similar effect on mice infected with a more virulent, mouse-adapted strain of influenza virus. Three-week-old male mice were fed Se-adequate or Se-deficient diet for 4 weeks before inoculation with influenza A/PR8/34. There was no difference in lung influenza viral titer between Se-deficient and Se-adequate mice. Se-deficient mice had less macrophage inflammatory protein 1α (MIP-1α) and regulated upon activation, normal T cell expressed and secreted (RANTES) production at the transcriptional and protein level in the lung postinfection. Se-deficient mice also had higher levels of IL-2 expression followed by a higher level of IL-4 expression in the lung. At Day 7 postinfection, there was no death in the Se-deficient group compared with 50% of the mice dying in the Se-adequate group. Sequencing of the virus isolated from infected Se-adequate and Se-deficient mice did not detect viral genome mutations in either group. This study demonstrated that Se-deficient mice had an altered immune response to an infection with a virulent strain of influenza virus. This altered immune response was beneficial for protecting the mice from influenza virus-induced mortality.
Introduction
Selenium (Se) is of fundamental importance to human health. It is an essential component of several major metabolic pathways, including thyroid hormone metabolism and antioxidant enzyme defense systems. Se is incorporated as selenocysteine at the active site of a wide range of selenoproteins. Under physiological conditions, the Se in selenocysteine is an extremely efficient biological catalyst. Among the selenoproteins with identified biological functions are the antioxidant enzymes glutathione peroxidase (GPX) and thioredoxin reductase (TrxR).
Dietary Se is essential for a healthy immune system (1) and Se influences both the innate and the adaptive immune responses (2, 3). The effects of Se deficiency include reduced T-cell numbers and impaired lymphocyte proliferation and function (4). Se supplementation enhances T-cell responses, stimulates antibody production and protects immune cells from oxidative-induced damage. The diverse effects of Se on immune function have been previously reviewed (1).
Infection with influenza virus causes a great deal of morbidity and mortality worldwide each year. In the United States alone, influenza virus infection results in over 36,000 deaths and 114,000 hospitalizations per year (5). Infection with influenza virus causes damage to both the lungs and airways because of inflammatory responses. Although the immune response is critical for the recovery from viral infection, it is also responsible for the lung inflammation that contributes significantly to lung pathology.
Previous work in our laboratory demonstrated that mice deficient in Se, which led to a decrease in the Se-containing enzyme GPX, were much more susceptible to infection with a mild influenza virus. Specifically, Se-deficient mice infected with a nonmouse-adapted strain of influenza virus, influenza A/Bangkok/1/79, developed much more severe lung pathology compared with Se-adequate mice (6). Immune function was altered in the infected Se-deficient mice, and the viral genome had changed in Se-deficient animals to a more virulent genotype. Once these changes occurred, even mice with normal Se status would develop severe pathology when infected with the newly mutated influenza virus (7). Because influenza A/Bangkok/1/79 is a human influenza virus strain that was not adapted to grow efficiently in mice, we questioned how a mouse-adapted, virulent strain of influenza virus would behave in a Se-deficient animal.
Materials and Methods
Influenza Virus.
Influenza A/Puerto Rico/8/34 (PR8) was propagated in 10-day-old, embryonated hen’s eggs (8). The virus-containing allantoic fluid was collected and stored at −80°C. This mouse-adapted strain of influenza virus causes a strong inflammatory response in normal mice (9).
Mice.
Three-week-old male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were housed four to a cage in the University of North Carolina at Chapel Hill (UNC-CH) animal facility, which is fully accredited by the American Association of Laboratory Animal Care. All mice were maintained under protocols approved by the Institutional Animal Use and Care Committee of the University of North Carolina at Chapel Hill. For all experiments, mice were provided with Se-adequate or Se-deficient diets for 4 weeks before inoculation with PR8. At baseline and various time points following infection, mice were killed, and the tissues were collected for the determination of lung pathology, liver GPX activity and glutathione (GS) levels, and lung proinflammatory chemokine and cytokine levels.
Diet.
The diet was obtained from Harlan (Indianapolis, IN). Se was added to the Se-adequate diet as sodium selenite. The Se content of the experimental diets was determined to be 200 ± 8 μg Se/kg for the Se-adequate diet and below the instrumental detection limit of 2.7 μg Se/kg for the Se-deficient diet.
Infection of Mice.
Mice were lightly anesthetized with an intraperitoneally (ip) injection of ketamine (0.022 mg) and xylazine (0.0156 mg). Following anesthesia, mice were infected intranasally with 2.5 plaque-forming unit (pfu) of PR8 in 0.05 ml sterile phosphate-buffered saline (PBS).
GPX Activity.
Liver GPX activity was determined according to the method of Paglia and Valentine (10). Briefly, liver was homogenated in 4× volume of sodium/ potassium (Na/K) phosphate buffer. Master mix (10 parts 0.4 M Na phosphate buffer, 5 parts sodium azide [NaN3], 4 parts doubled-distilled, deionized water [dddH2O]), 5 mM reduced glutathione [GSH] and 9 μl of glutathione reductase (GR), and 5 μl liver homogenate were combined. After blanking, 80 μl of 6 mM nicotinamide adenine dinucleotide phosphate (NADPH) was added to each sample. Samples were incubated for 1 min at 37°C, followed by addition of 50 μl hydrogen peroxide (H2O2). Absorption at A340 was immediately measured for 1 min at 20-sec intervals. One milliunit of enzyme activity was defined as 1 nM of NADPH oxidized to NADP per mg of protein per min.
GS and GSH Analysis.
Total GS and GSH were analyzed in tissue extracts prepared in 5% 5-sulfosalicylic acid (S2130, Sigma Chemical Co., St. Louis, MO) using a GR-coupled recycling assay (11). GSH disulfide (GSSG) concentrations were determined in extracts pretreated with 2-vinylpyridine (132292, Sigma). GSH was determined by subtracting GSSG from total GS.
Histopathology of Lungs.
The left lung was removed and inflated with 4% paraformaldehyde in 0.1 M of Na-phosphate buffer (pH 7.2). Sections (6 μm) were fixed in acetone and stained with hematoxylin-eosin. The extent of inflammation was graded without knowledge of the experimental variables by two independent investigators. Grading was performed semiquantitatively according to the relative degree (from lung to lung) of inflammatory infiltration. The scoring was as follows: 0, no inflammation; 1+, mild influx of inflammatory cells with inflammatory infiltrates clustered around vessels; 2+, increased inflammation with approximately 25%–50% of the total lung involved; 3+, severe inflammation involving 50%–70% of the lung; and 4+, almost all lung tissue contains inflammatory infiltrates.
Quantitation of Viral Titer by Real-Time Polymerase Chain Reaction.
To determine lung viral titers, half of the right lung was removed, and total RNA was isolated using the TRIzol method (Invitrogen, Carlsbad, CA). Reverse transcription was carried out using Superscript II First Strand Synthesis kit (11904–018, Invitrogen) using random hexamer primers. Expression of the influenza matrix (M1) gene and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were determined by quantitative real-time PCR (qRT-PCR) as described (12). Fluorescent reporters were detected using Bio-Rad (Hercules, CA) iCycler PCR machine and primers and probes were obtained from Applied Biosystems (Foster City, CA). The levels of mRNA for GAPDH were determined for all samples and were used to normalize expression of the influenza M1 gene. Data were converted to hemagglutination units (HAU) using real-time PCR standards made from the virus stock with known HAU titer.
Quantitation of Lung Chemokine and Cytokine mRNA by Real-Time PCR.
mRNA levels for regulated upon activation, normal T cell expressed and secreted (RANTES), murine macrophage inflammatory protein 1α (MIP-1α), interleukin (IL)-2, IL-4, and GAPDH were determined using qRT-PCR, as described above. Because there was no statistical difference in mRNA levels between Se-adequate and Se-deficient groups at Day 0 (uninfected), data were expressed as the ratio to the Day 0 levels of the Se-adequate group.
Quantitation of Lung Chemokine Protein by Enzyme-Linked Immunoabsorbent Assay (ELI-SA).
Half of the right lung was removed and homogenized in 1 mL PBS. Measurement of RANTES and MIP-1α were performed with ELISA kits (DY478, DY450, R&D Systems, Minneapolis, MN) according to protocols provided by the manufacturer. Results were normalized to the amount of total protein.
Virus Sequencing.
Viral RNA isolation and reverse transcription were carried out as described above. Primers were designed for amplification of the matrix (M) and hemagglutinin (HA) genes. PCR was performed with the following primer sets: M, 5′-GATGAGTCTTCTAACC-GAGGT-3′ and 3′-AAACAGTCGTATCTCGACCTCA-5′; and HA, 5′-TGAAGGCAAACCTACTGGTCC-3′ and 3′-ACCTAGAAACGTCACGTCTT-5′. PCR products were purified using the QIA quick PCR purification kit (Qiagen, Valencia, CA). DNA was sequenced at the UNC-CH Automated DNA Sequencing Facility on a Model 377 DNA sequencer (Applied Biosystems Division, Perkin Elmer, Boston, MA) by using the ABI Prism Dye Terminator Cycle Sequencing Ready Reaction Kit with AmpliTaq DNA Polymerase (Applied Biosystems Division, Perkin Elmer). Sequencing data were analyzed with Sequencher 4.5 (Gene Codes Corporation, Ann Arbor, MI).
Statistical Analysis.
Lung pathology, GPX activity, and glutathione data were analyzed by two-way ANOVA followed by Tukey HSD test. Real-time PCR and ELISA data were analyzed by Kruskal-Wallis test. All statistical analyses were performed with JMP software (SAS, Cary, NC).
Results
Se-Deficient Mice Had Decreased GPX Activity.
Before influenza virus infection (Day 0) and at 3 and 7 days postinfection, mice fed the Se-deficient diet had significantly lower levels of GPX activity in the liver compared with mice fed the Se-adequate diet, indicating that Se deficiency occurred in the mice on the deficient diet. Infection induced an increase in GPX activity in the Se-adequate mice at Day 3 postinfection compared with the Se-adequate mice at Day 0 (Fig. 1). The increased GPX activity in infected Se-adequate mice suggests that these mice increased the amount of GPX in response to the infection-induced oxidative stress, whereas Se-deficient mice were not able to do so because of the limited supply of Se, which is essential for the synthesis of GPX.
Se-Deficient Mice Had Less Amounts of GS and GSH.
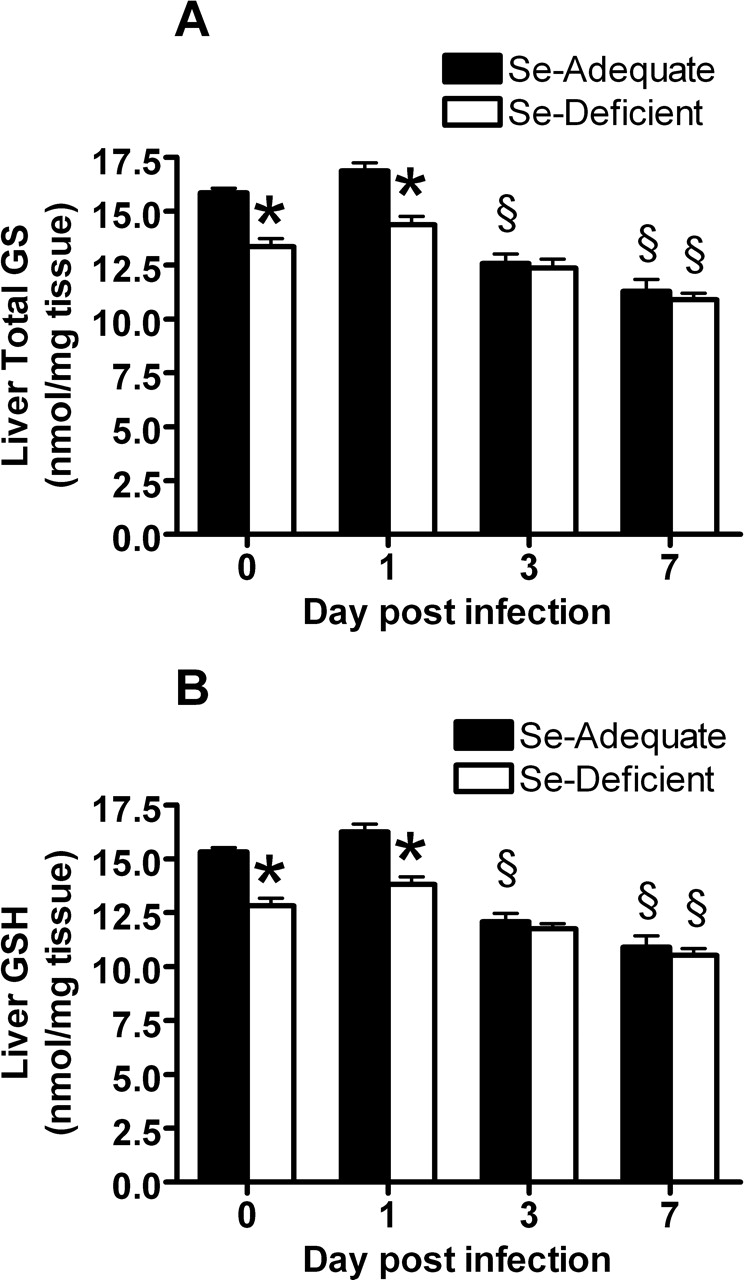
Before influenza virus infection (Day 0), Se-deficient mice had significantly lower levels of total GS (Fig. 2A) and GSH (Fig. 2B) in the liver compared with Se-adequate mice, suggesting that Se-deficient mice were oxidatively stressed before infection. Following infection, GS and GSH levels were significantly decreased at Day 7. Infection also induced a decrease in GS and GSH levels in Se-adequate mice at 3 and 7 days postinfection, suggesting that influenza infection induced oxidative stress in Se-adequate mice as well (Fig. 2A and B).
Lung Pathology Similar Between Se-Adequate and Se-Deficient Mice.
As shown in Figure 3, lung pathology was not statistically different between Se-adequate and Se-deficient mice at Days 3 and 6 post-infection. This is in contrast to our previous study with influenza A/Bangkok virus, in which Se-deficient mice had increased pathology.
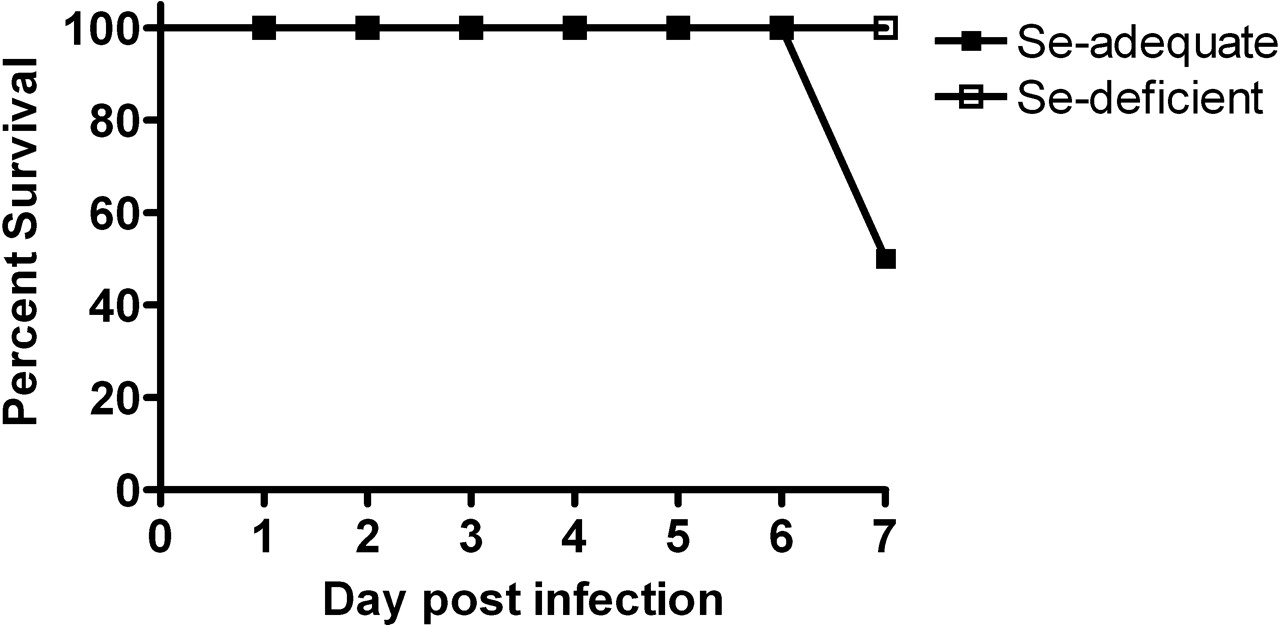
Se-Deficient Mice Had a Lower Influenza-Induced Mortality Rate.
At Day 7 postinfluenza virus infection, Se-adequate mice had a 50% mortality rate, whereas no mice in the Se-deficient group died (Fig. 4).
Lung Influenza Viral Titer.
Because of the higher mortality rate in the infected Se-adequate mice, we reasoned that lung influenza viral titers would be higher in Se-adequate animals. However, there were no differences in viral titer between Se-adequate and Se-deficient groups. Peak influenza viral titers occurred at Day 3 postinfection and decreased at Day 7 in both Se-adequate and Se-deficient mice (Fig. 5).
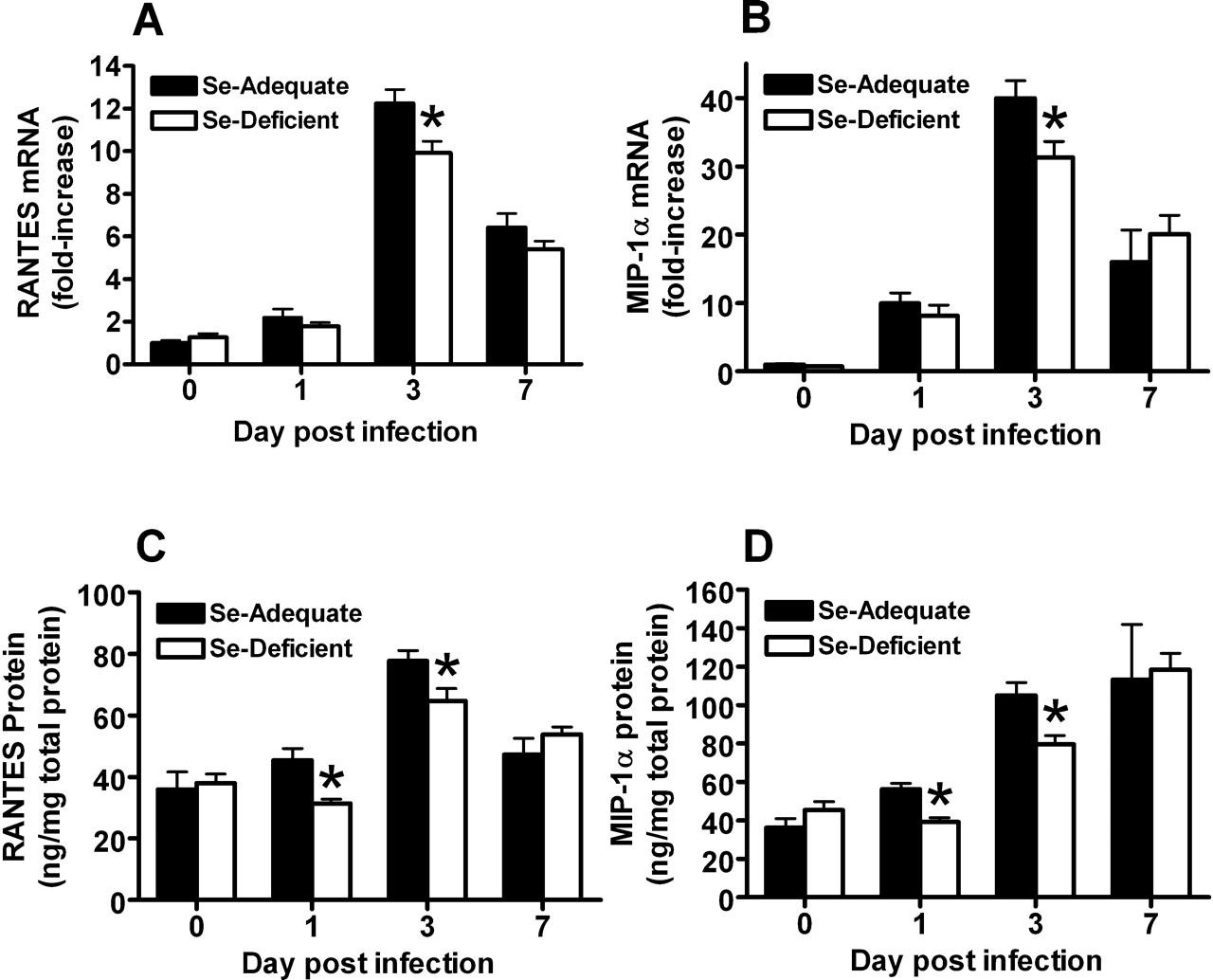
Se-Deficient Mice Had Lower RANTES and MIP-1α Levels in the Lung Postinfluenza Virus Infection.
Previous studies have demonstrated the importance of chemokines in the development of influenza-induced inflammation (13–15). To determine whether Se deficiency influenced chemokine production, both lung mRNA and protein levels for RANTES and MIP-1α and mRNA levels for monocyte chemotactic protein 1 (MCP-1) were measured. Chemokine mRNA levels for MCP-1 increased at 1, 3, and 7 days postinfection, but there was no difference in the levels between Se-adequate and Se-deficient mice (data not shown). However, although mRNA levels for RANTES and MIP-1α also increased postinfection, Se-deficient mice had lower RANTES and MIP-1α expression at Day 3 post-infection compared with Se-deficient mice (Fig. 6A and B). To confirm that mRNA levels correlated with protein levels, lung RANTES and MIP-1α protein levels were measured. Se-deficient mice had less amounts of RANTES and MIP-1α proteins at 1 and 3 days postinfection (Fig. 6C and D), correlating with the mRNA data.
Se-Deficient Mice Had Higher Levels of IL-2 Expression Followed by a Higher Level of IL-4 Expression in the Lung Postinfluenza Virus Infection.
Cytokine production in the lungs is also a hallmark characteristic of influenza infection. To determine whether Se deficiency could affect cytokine production, we measured lung mRNA for levels of tumor necrosis factor alpha (TNF-α), IL-1β, IL-12, IL-2, IL-4, interferon gamma (IFN-γ) and IL-10. Only IL-2 and IL-4 levels demonstrated significant differences between Se-adequate and Se-deficient mice (Fig. 7A and B). IL-2 mRNA levels were higher in Se-deficient mice at 1 and 3 days postinfection (Fig. 7A), and IL-4 mRNA level was higher at Day 7 postinfection in the Se-deficient mice compared with the Se-adequate controls (Fig. 7B). Se-deficient mice also had a trend toward a higher INF-γ mRNA production at Day 1 postinfection (P = 0.06, data not shown).
No Mutations Were Detected in Pr8 Viral Genome After Replicating in Se-Adequate or Se-Deficient Mice.
The M gene and HA gene of the PR8 viral genome isolated from Se-adequate and Se-deficient mice at Day 7 postinfection were sequenced and compared with the sequence of the virus stock used for infection. No mutations were detected.
Discussion
The fundamental importance of Se to optimal immune function has been widely demonstrated. An adequate Se intake is essential for an appropriate immune response to various infectious diseases. For example, Se supplementation results in more rapid poliovirus clearance (16) as well as lowered hospitalization rates among human immunodeficiency virus (HIV)-infected patients (17). Se deficiency is associated with Keshan disease, a disease correlated with coxsackievirus infection of the heart muscle (18). By using a murine model of coxsackievirus B3 (CVB3)-induced myocarditis, our laboratory demonstrated that Se-deficient mice were more susceptible to the cardiopathologic effects of the virus. In addition, a normally benign strain of CVB3 becomes virulent in Se-deficient mice because of changes in the viral genome (19). Previous work in our laboratory also demonstrated that Se-deficient mice were more susceptible to infection with mild influenza virus. Specifically, Se-deficient mice infected with a mild strain of influenza virus, influenza A/Bangkok/1/79, developed much more severe lung pathology compared with Se-adequate mice. Immune function was altered in the infected Se-deficient mice, and the viral genome had changed in the deficient animals to a more virulent genotype (6, 7).
The strain of influenza virus used in these previous studies, influenza A/Bangkok/1/79, is a human strain that induces a mild inflammatory response in normal mice. In this study, a mouse-adapted strain of influenza virus, PR8, was used. This strain of virus induces a much more severe lung pathology compared with the nonmouse-adapted Bangkok strain. Both Se-adequate and Se-deficient mice had severe lung inflammation following infection with PR8. Because PR8 induces a stronger inflammatory response in the lungs compared with the Bangkok strain, it was not surprising that pathology differences between Se-adequate and Se-deficient mice were not seen. What was of interest, however, was that PR8-infected Se-deficient mice demonstrated no mortality at Day 7 compared with Se-adequate mice, which had a 50% mortality rate. This result suggested that the lung pathology continued to increase in the Se-adequate mice, resulting in death, but the pathology did not accelerate as rapidly in the Se-deficient mice. To determine the reason for the lower mortality and lung pathology in the Se-deficient mice, we measured lung viral titers. Milder lung pathology may have been related to lower lung viral titers in the Se-deficient mice. However, no differences in the lung influenza viral titers were found between Se-adequate and Se-deficient mice, suggesting that Se levels did not influence the ability of the host to eradicate the virus. This result was similar to our study with Bangkok virus, in which viral titers were equivalent between Se-adequate and Se-deficient mice.
Another possibility for the difference in lung pathology may be in the production of lung chemokines and cytokines, which are responsible for generating the lung inflammation postinfection. We found that influenza-infected Se-deficient mice had less production of the chemokines RANTES and MIP-1α in the lungs compared with Se-adequate mice. Chemokines are potent chemoattractant cytokines and have been considered the main candidate molecules responsible for the selective recruitment of distinct leukocyte populations. RANTES is produced by CD8+ T cells, epithelial cells, fibroblasts, and platelets and plays a key role in the immune response to viral infection (20). MIP-1α is produced by a variety of cell types, including monocytes, macrophages, mast cells, Langerhans cells, fibroblasts, and T cells. MIP-1α is primarily chemotactic for B cells, activated CD8+ T cells, natural killer (NK) cells, and eosinophils (21–24). MIP-1α also increases cell adhesion by inducing intercellular adhesion molecule-1 (ICAM-1) expression (25).
Influenza virus–infected MIP-1α-knockout mice have significantly less lung inflammation compared with MIP-1α wild-type controls, suggesting that MIP-1α plays a critical role in the inflammatory response to influenza virus infection (13). Because chemokines are important mediators of inflammation, the lower levels of RANTES and MIP-1α in the lungs of the Se-deficient mice may have prevented the severe lung inflammation that caused mortality in the Se-adequate mice.
Infection with influenza virus induces a strong T-helper type 1 (Th1) response, resulting in expansion of influenza-specific CD8+ T cells, which play a significant role in viral clearance. However, the Se-deficient mice had a more T-helper type 2 (Th2)-like pattern of cytokine expression. IL-4 is an important mediator of Th2 type responses, and the IL-4 expression was much higher in the lungs of the Se-deficient mice at Day 7 postinfection. IL-2 mRNA levels were also higher in the lungs of the Se-deficient mice at 1 and 3 days postinfection compared with the Se-adequate controls. IL-2 stimulates T-cell activation and expansion (26, 27), especially the development of Th2 cells by stabilizing the accessibility of the IL-4 gene (28). In vivo, IL-2 neutralization inhibits IL-4 production (28). Thus, the higher mRNA level of IL-4 at Day 7 may have been stimulated by the earlier higher levels of IL-2 in Se-deficient mice. In addition, RANTES and MIP-1α have been correlated with a Th1 response (29–32), and MIP-1α drives the development of Th1 cells in vitro (33), again suggesting that the lower levels of these chemokines in the Se-deficient mice skewed the response toward a Th2 rather than a Th1 response.
Why does Se deficiency favor a Th2 response? The lower GSH levels in Se-deficient mice may be a possible explanation. GSH levels in the immune cells play a pivotal role in determining the Th1/Th2 balance. GSH depletion in antigen presenting cells (APCs) shifts the immune response toward a Th2 response both in vitro and in vivo (34). The intracellular GSH levels have also been correlated with the Th1 cytokine production versus the Th2 cytokine production by macrophages and CD4+ T cells (35). The effect of GSH levels on Th1/Th2 balance is partly mediated by IL-12 and IL-4 (34, 35). Our data compliment these studies by showing a higher level of IL-4 expression in Se-deficient mice. Although a difference in IL-12 expression between Se-adequate and Se-deficient mice was not observed in our study, the diminished Th1 response in Se-deficient mice may be explained by the changes in production of other chemokines and cytokines by APCs and macrophages, such as RANTES, MIP-1, and possibly, IL-18.
In addition, effects of Se deficiency on immune function may not be solely explained by the change in GSH levels. Approximately 25 selenoproteins have been identified, many with unknown biological function (36). It is possible some of these selenoproteins have immunomodulatory functions. With the progression of our understanding of the function of selenoproteins, more detailed mechanisms will be elucidated.
Previous studies in our laboratory had demonstrated that influenza A/Bangkok/1/79, a mild strain of influenza virus in mice, underwent substantial genetic mutations in viral M gene, after replicating in Se-deficient mice, which turned the virus into a more virulent strain (7). However, in the current study with PR8 infection, no mutations were detected in this gene. We also sequenced the viral HA gene, a viral RNA segment with a high mutation rate, and found no mutations. As a human strain of influenza virus, influenza A/Bangkok/1/79 does not replicate efficiently in mice under normal conditions. In contrast, PR8 is a mouse-adapted strain of influenza and replicates well in mice under normal conditions. Thus, less host-adapted viruses may be more susceptible to mutations induced by a nutritional deficiency.
The lower mortality rate in Se-deficient mice was not caused by a lower viral titer, as Se-adequate mice and Se-deficient mice had similar viral titers in the lung. Instead, the lower mortality was a result of the reduced inflammatory response in these mice. Although the reduced immune response increased the survival of the Se-deficient mice, this may not be beneficial for an infection in which the inflammatory response plays a more critical role in viral eradication. Thus, a careful balance between the inflammation required for virus control versus the inflammation that damages tissues must be struck. In this study, Se deficiency tipped the balance in favor of reduced inflammation, which proved to be beneficial to the animals.
In summary, Se-deficient mice had an altered immune response to influenza virus infection, which was characterized by a diminished Th1 response and an enhanced Th2 response. Less production of chemokines in the lungs of Se-deficient mice contributed to less lung pathology and higher survival rates. Our results suggest that the pathological outcome of a virus infection of Se-deficient mice is dependent not only on Se status, but also the virulence of the infecting viral strain as well. Further studies to investigate the influence of Se on immune function are warranted.
GPX activity in the liver before influenza virus infection (Day 0), and at 3 and 7 days postinfection. Mice were fed the Se-adequate or Se-deficient diets for 4 weeks before infection. Data are expressed as mean ± SEM, n = 5. Asterisks indicate significant difference between the Se-adequate and Se-deficient groups (P < 0.001). § indicates significant difference between the level post-infection and the level before infection in either the Se-adequate group or Se-deficient group (P < 0.05). (A) GS and (B) GSH in the liver before influenza virus infection (Day 0) and at 1, 3, and 7 days postinfection. Mice were fed the Se-adequate or Se-deficient diets for 4 weeks before infection. Data are expressed as mean ± SEM, n = 8. Asterisks indicate significant difference between Se-adequate and Se-deficient groups (P < 0.01). § indicates significant difference between the level postinfection and the level before infection in either the Se-adequate group or Se-deficient group (P < 0.01). Histopathologic scores of influenza infected Se-adequate and Se-deficient mice Day 3 and 6 postinoculation. Data are expressed as mean ± SEM, n = 8. No significant differences between groups. Survival rate postinfluenza virus infection in Se-adequate and Se-deficient mice. Mice were fed the Se-adequate or Se-deficient diets for 4 weeks before infection. Data are expressed as percentage of survival, n = 8. Lung influenza viral titers before influenza virus infection (Day 0) and at 1, 3, and 7 days postinfection. Mice were fed the Se-adequate or Se-deficient diets for 4 weeks before infection. Data are expressed as mean ± SEM, n = 8. (A, B) Lung mRNA (C, D) and protein levels for RANTES and MIP-1α from Se-adequate or Se-deficient mice before influenza virus infection (Day 0) and at 1, 3, and 7 days postinfection. mRNA data are expressed as the ratio to the Day 0 levels of the Se-adequate group ± SEM. There were no significant differences of Day 0 mRNA levels between Se-adequate and Se-deficient mice. Protein data are expressed as ng RANTES or MIP-1α/mg total tissue protein ± SEM, n = 8. Asterisks indicate significant difference between Se-adequate and Se-deficient groups (P < 0.05). (A) Lung mRNA levels for IL-2 and (B) IL-4 from Se-adequate or Se-deficient mice before influenza virus infection (Day 0) and at 1, 3, and 7 days postinfection. Data are expressed as the ratio to the Day 0 levels of the Se-adequate group ± SEM. There were no significant differences of Day 0 mRNA levels between Se-adequate and Se-deficient mice, n = 8. Asterisks indicate significant difference between Se-adequate and Se-deficient groups (P < 0.05).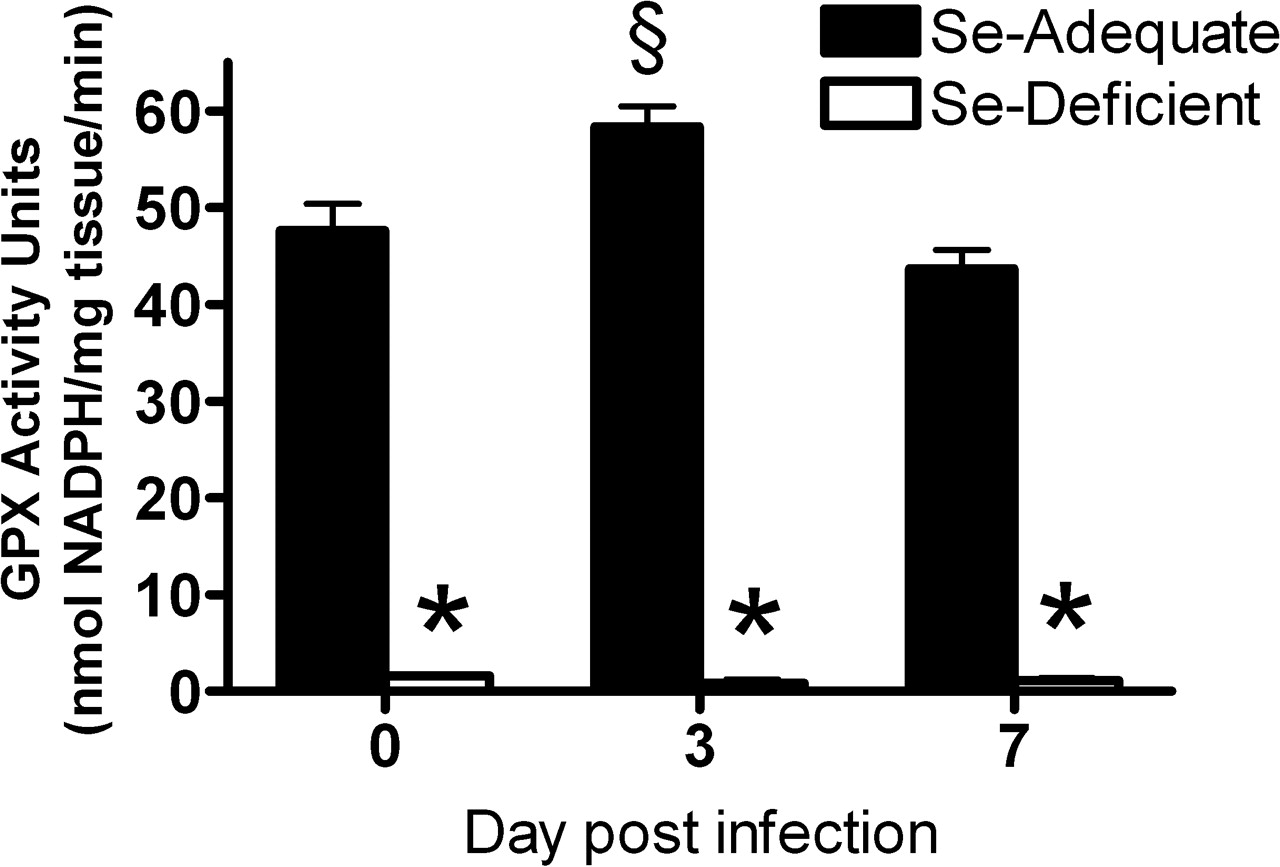



Footnotes
This work was supported by grant AI055050 from the National Institutes of Health and by grant DK56350 from the National Institutes of Health, which funded the Clinical Nutrition Research Unit.