
Abstract
Allelic variation in the mouse beta globin gene complex (Hbb) produces structurally different beta globins in different mouse strains. Like humans, mice with HbbS alleles produce a single beta globin with one reactive cysteine (β Cys93). In contrast, mice with HbbD alleles produce two structurally different beta globins, each containing an additional cysteine (β Cys13). β Cys93 forms mixed disulfides with glutathione and plays a pivotal role in the activities of hemoglobin, glutathione, and nitric oxide. Similar roles for mouse β Cys13 have not been described. We used capillary electrophoresis to compare reduced glutathione (GSH), glutathione disulfide (GSSG), and S-glutathionyl hemoglobin levels in erythrocytes from inbred C57BL/6J (homozygous HbbS/S) and 129S1/SvImJ (homozygous HbbD/D) mice and their homozygous and heterozygous B6129S/F2J hybrid offspring. S-glutathionyl hemoglobin was nearly undetectable in inbred or hybrid mice with only monocysteinyl beta globins (HbbS/S) but represented up to 10% of total hemoglobin in mice with polycysteinyl beta globins (HbbS/D or HbbD/D). The stepwise increase in beta globin sulfhydryl group concentration in HbbS/S, HbbS/D, and HbbD/D F2 mice was associated with increasing hemoglobin-bound glutathione and decreasing free glutathione (GSH + GSSG) concentrations. Total erythrocyte glutathione (GSH + GSSG + hemoglobin-bound) was not significantly different between groups. In vitro studies showed that β Cys13 in mouse HbbD beta globins was more susceptible to disulfide exchange with GSSG than β Cys93. We conclude that reactive beta globin sulfhydryl group concentration is genetically determined in mice, and that polycysteinyl beta globins markedly influence intraerythrocyte glutathione distribution between free and hemoglobin-bound compartments. Although Hbb heterozygosity and polycysteinyl beta globins are common in wild mouse populations, all common human beta globins contain only one reactive cysteine, and homozygosity is the norm. These fundamental differences in mouse and human beta globin genetics have important implications for the study of mouse biology and for the use of some mouse strains as models for humans.
Introduction
Unlike humans, mice inherit a pair of linked beta globin genes from each parent. Three major beta globin gene complex (Hbb) haplotypes are common in wild and inbred mice: HbbS, HbbD, and HbbP (1–3). Mice with the HbbS haplotype (e.g., C57BL/6J) have two similar alleles at both beta globin loci. These produce structurally identical beta globins (β S) that migrate during electrophoresis in a dimer with alpha globin as a single band (hence the S designation; Ref. 4). Mice with the HbbD (e.g., 129S1/SvImJ) or HbbP haplotypes have two different beta globin alleles. These produce beta globins that are structurally different from each other and from the beta globin produced by HbbS mice. Beta globins produced by HbbD mice (β Dmajor and β Dminor) migrate during electrophoresis in a dimer with alpha globin as diffuse or double bands (hence the D designation). Because the migration patterns of hemoglobins from HbbD and HbbP mice are similar, the D designation was originally applied to both haplotypes until allelic variation was identified by protein sequencing (5, 6).
The most prevalent mouse beta globins are identified as β S in mice with the HbbS haplotype, β Dmajor and β Dminor in mice with the HbbD haplotype, and β Pmajor and β Pminor in mice with the HbbP haplotype (although β Dmajor and β Pmajor are structurally identical; Ref. 7). Both β Dminor and β Pminor are expressed at low levels compared with their major counterparts (~20% of total beta globin in homozygous mice), hence the minor designation. One way that β S is similar to human beta globin but differs from all other common mouse beta globins is the presence of a single reactive cysteine residue at position 93 (β Cys93; Fig. 1). In contrast, mice with HbbD or HbbP haplotypes have an additional cysteine residue (β Cys13) in both their major and minor beta globin variants.
The sulfhydryl group of β Cys93 plays important roles in nitric oxide (NO) metabolism (8) and in the intermolecular formation of mixed disulfides with other sulfhydryl proteins and glutathione (9, 10). Similar roles for mouse β Cys13 have not been reported, but phenotypic differences between HbbS and HbbD mice have been noted. Kruckebeg et al. (11) reported that erythrocytes from HbbD mice were more susceptible to hydrogen peroxide–induced hemolysis compared with HbbS mice. Newton and Peters (12) reported that hemoglobin from HbbD mice had higher oxygen affinity than hemoglobin from HbbS mice.
Preliminary studies in our laboratory detected high levels of what we suspected was S-glutathionyl hemoglobin in HbbD but not HbbS mice using high-resolution capillary isoelectric focusing (cIEF). S-glutathionyl hemoglobin is formed under oxidative conditions (13). Its use as a clinical marker of oxidative stress in humans has been proposed (14). The purpose of these experiments was to confirm the identity of mouse hemoglobin variants observed on cIEF and assess the relationship between mouse Hbb haplotype and erythrocyte glutathione metabolism.
Materials and Methods
All reagents were obtained from Sigma Chemical Co. (St. Louis, MO) unless otherwise noted. Six-week-old male inbred C57BL/6J mice (C57; n = 10), inbred 129S1/SvImJ mice (129; n = 10), and hybrid B6129S/F2J mice (F2; n = 40) were obtained from Jackson Laboratory (Bar Harbor, ME). Use of animals in this project was approved by the Research Institute for Children Institutional Animal Care and Use Committee. The mice were group housed under conventional environmental conditions and received laboratory chow and tap water ad libitum for the duration of the experiment. After 2 weeks of acclimatization the nonfasted mice were weighed, and blood was collected from the tail for hemoglobin variant analysis and quantification of reduced glutathione (GSH) and glutathione disulfide (GSSG). The GenBank numbers for the mouse beta globins are: HbbS, NP_032246; and HbbD, P02088.
Hemolysates were prepared for hemoglobin variant analysis by adding 10 μ l erythrocytes to 200 μ l hemolyzing reagent (10 mM KCN, 5 mM EDTA). Structural and post-translationally modified mouse hemoglobin variants were identified and quantified by capillary isoelectric focusing (cIEF) as previously described for human hemoglobins (15–18), except we used 80 mM boric acid (pH 10.2) as the cathode buffer and narrower-range ampholytes (pH 6.7 to 7.7). Absorbance was monitored at 415 nm (specific for heme groups), and the proportions of different mouse hemoglobin variants were expressed as percentage of total hemoglobin based on peak areas. The isoelectric points (pIs) of mouse hemoglobin variants were determined by cIEF analysis of mouse hemolysates mixed with human hemolysate containing hemoglobin variants with known pIs (HbA2, 7.41; HbS, 7.21; HbF, 7.06; HbA, 6.97; and HbA1c, 6.94). S-glutathionyl hemoglobin was synthesized in vitro by disulfide exchange with GSSG (19). A stock solution (100 mM GSSG) was prepared in deionized water, then diluted with water as needed to prepare working solutions with different GSSG concentrations. Working GSSG solutions (10 μ l) were then added to 90 μ l hemolysate prepared as described above, and the mixtures were incubated for varying lengths of time (up to 24 hrs) at 37° C.
GSH and GSSG were simultaneously measured (μ M erythrocytes) by capillary zone electrophoresis following extraction from packed erythrocytes with metaphosphoric acid using a modification of the assay described by Serru et al. (20) as previously described (21). The molar concentration of hemoglobin-bound glutathione was estimated based on percent total S-glutathionyl hemoglobin using 4261 micromoles hemoglobin per liter erythrocytes (derived from average mouse hematocrit and blood hemoglobin concentrations) and 1 mole glutathione per mole S-glutathionyl hemoglobin as constants. Total erythrocyte glutathione (μ M erythrocytes) was then calculated as the sum of μ M GSH + (2 × μ M GSSG) + μ M hemoglobin-bound glutathione.
Results were analyzed using PC-SAS version 9.1 (SAS Institute, Cary, NC). Statistical treatment groups included the two parent strains and the three groups of F2 mice classified by Hbb haplotype. All measurement data were tested for normality (Shapiro-Wilk test) and variance homogeneity (Levene’s test) prior to one-way ANOVA. Values for all variables were approximately normally distributed (P > 0.01). Variables that failed the homogeneity of variance test were analyzed using Welch’s ANOVA, which is robust to the homogeneity of variance assumption. Variables with significant ANOVA results were further analyzed by post hoc multiple pair-wise comparisons (pair-wise t test or least significant difference test) to determine which group means were significantly different. Unless otherwise noted, differences between groups were considered significant at α < 0.05.
Results
Capillary Isoelectric Focusing of Mouse Hemoglobin Variants.
To interpret cIEF electropherograms, it is important to note that although hemoglobin is a tetramer consisting of two alpha and two beta globins, globin chains migrate as α β dimers when analyzed by most charge-based separation techniques (9). In cIEF, positively charged structural or post-translationally modified α β dimers have higher pIs and elute earlier than more negatively charged α β dimers. Disulfide formation with glutathione introduces the glutamyl carboxyl group of glutathione into the beta globin molecule, which adds a negative charge to the α β dimer and lowers its pI.
Figure 2 shows the cIEF patterns of mouse hemoglobin variants present in erythrocyte hemolysates from 129 and C57 mice incubated with or without GSSG. Untreated hemolysates from 129 mice (top left panel) exhibited two relatively abundant cIEF peaks and two smaller peaks with lower pIs (higher migration time). Based on the expected structural hemoglobin variants in HbbD/D mice and their reported proportions and electrophoretic properties, peak 1 is α β Dminor and peak 2 is α β Dmajor; the higher pI of α β Dminor is likely due to an Asn→Lys substitution at position 76 (Fig. 1). Identification of peaks 3 and 4 as mono-S-glutathionyl adducts of α β Dminor and α β Dmajor, respectively, is based in part on the fact that the size of these peaks can be decreased by reduction with mercaptoethanol (Fig. 3). In contrast, untreated hemolysates from C57 mice (Fig. 2, top right panel) exhibited a single abundant hemoglobin peak with a pI and migration time similar to that of peak 2 in 129 mice. Identification of peak 2 in C57 mice as α β S is based on the fact that β S is the only structural beta globin variant present in homozygous HbbS/S mice. The small peak adjacent to peak 2 contains glycated α β S (22).
Synthesis of S-glutathionyl hemoglobins in vitro by disulfide exchange with GSSG provided further proof of the chemical compositions of peaks 3 and 4 in 129 mice. Figure 2 shows that incubation of hemolysates from 129 mice with GSSG for 5 hrs (bottom left panel) caused the nearly complete disappearance of peaks 1 and 2, a marked increase in the sizes of peaks 3 and 4, and the appearance of two new peaks with very low pIs (peaks 5 and 6) that were not detected in untreated hemolysates. The lower pIs of peaks 3 and 4 compared with peaks 1 and 2 are attributable to the addition of a single negatively charged glutathione molecule to α β Dminor and α β Dmajor, respectively, forming mono-S-glutathionyl α β dimers. The even lower pIs of peaks 5 and 6 are attributable to the addition of another negatively charged glutathione molecule forming di-S-glutathionyl α β Dminor and α β Dmajor, respectively. These results clearly show that disulfide exchange with GSSG occurs at both beta globin cysteine sulfhydryl groups in both β Dminor and β Dmajor.
In contrast, hemolysates from C57 mice incubated with GSSG (bottom right panel) have only one additional low pI peak (peak 7). The appearance of peak 7 coincided with a reduction in the area of peak 2 and is explained by the addition of a single negatively charged glutathione molecule to α β S by disulfide exchange. Since β Cys93 is the only reactive cysteine present in β S, peak 7 is S-glutathionyl-β Cys93 α β S. Incubating hemolysates at different GSSG concentrations for different lengths of time showed that β Cys13 was more reactive than β Cys93. Peaks 3 and 4 in hemolysates from 129 mice rapidly increased in size even at relatively low GSSG concentrations (data not shown). Only when peaks 1 and 2 were nearly completely depleted did peaks 5 and 6 begin to appear. Peak 7 (C57 mice) and peaks 5 and 6 (129 mice) formed at similarly slow rates in 5 mM GSSG, suggesting that the involved sulfhydryl group is the same in each case. We therefore conclude that rapidly forming peaks 3 and 4 are mono-S-glutathionyl-β Cys13 α β Dminor and α β Dmajor, respectively. Slow-forming peaks 5 and 6 are di-S-glutathionyl-β Cys13/β Cys93 α β Dminor and α β Dmajor, respectively. It is important to emphasize that di-S-glutathionyl α β Dminor and α β Dmajor were not detected in untreated hemolysates.
Hbb Haplotype and Intraerythrocyte Glutathione Distribution in Inbred and F2 Hybrid Mice.
The Hbb haplotypes of the 40 F2 hybrid mice were determined based on their hemoglobin variant phenotypes compared with the parent strains. Figure 4 is a magnified view of the absorbance profiles of hemolysates from F2 hybrid mice with different Hbb haplotypes. Ten F2 mice had cIEF profiles similar to those of the inbred C57 mice (homozygous HbbS/S), and 11 had profiles similar to those of the inbred 129 mice (homozygous HbbD/D). The remaining 19 F2 mice (heterozygous HbbS/D) had profiles similar to those of the 129 mice, but with only half as much α β Dminor (peak 1), S-glutathionyl α β Dminor (peak 3) and S-glutathionyl α β Dmajor (peak 4; Table1). Peak 2 in the HbbS/D F2 mice contains both α β S and α β Dmajor, which have similar pIs and are indistinguishable under these cIEF conditions. The distribution of Hbb alleles in the F2 mice approximates that expected by Mendelian inheritance.
Table 1 shows that Hbb haplotype was strongly associated with intraerythrocyte glutathione distribution. GSH and GSSG levels were significantly higher in C57 mice compared with 129 mice, but hemoglobin-bound glutathione levels were approximately 8-fold lower. GSH and GSSG levels in F2 mice with homozygous HbbS/S or HbbD/D haplotypes were not significantly different from those of inbred mice with the same Hbb haplotype. F2 mice with the heterozygous HbbS/D haplotype had mean values for GSH, GSSG, and hemoglobin-bound glutathione that were intermediate to those of the homozygous F2 mice. Total erythrocyte glutathione (sum of GSH, GSSG, and hemoglobin-bound) was not significantly different between groups. The molar distribution of glutathione between free (GSH + GSSG) and hemoglobin-bound compartments was: 97.7% and 2.3% (C57, HbbS/S); 97.5% and 2.5% (F2, HbbS/S); 88.7% and 11.3% (F2, HbbS/D); 83.7% and 16.3% (F2, HbbD/D); and 79.8% and 20.2% (129, HbbD/D).
Discussion
Allelic variation in the mouse beta globin gene complex was first detected over 50 years ago based on the chromatographic and electrophoretic heterogeneity of mouse hemoglobin variants (23, 24). Globin sequencing later uncovered the existence of monocysteinyl and dicysteinyl mouse beta globins (5), but the biologic implications of polycysteinyl beta globins appear to have been overlooked. Our results show that both β Cys13 and β Cys93 can be modified by glutathione disulfide and that β Cys13 is more susceptible to modification than β Cys93. The concentrations of GSSG used in the in vitro experiments were above physiologic, but higher reactivity of β Cys13 under physiologic conditions is supported by the presence of naturally occurring S-glutathionyl-β Cys13 α β Dminor and α β Dmajor in untreated hemolysates from heterozygous HbbS/D or homozygous HbbD/D mice.
Analysis of erythrocyte glutathione showed a direct link between Hbb haplotype, beta globin cysteine sulfhydryl group concentration, and glutathione metabolism in vivo. Since β Cys93 is conserved in all mouse and human beta globins, all mice and humans have similar amounts of β Cys93 (i.e., 1.0 mole β Cys93 per mole beta globin). In contrast, the erythrocyte concentration of β Cys13 is variable in hybrid mice, ranging from zero in homozygous HbbS/S mice to 0.5 and 1.0 moles β Cys13 per mole beta globin in heterozygous HbbS/D and homozygous HbbD/D mice, respectively. This genetically determined stepwise increase in beta globin sulfhydryl group concentration was associated with a stepwise increase in hemoglobin-bound glutathione and a proportional decrease in free glutathione. Total erythrocyte glutathione concentration was not significantly different between groups.
Although the F2 mice used in these experiments had mixed C57/129 genetic backgrounds, parental alleles should be randomly distributed at non-Hbb loci, and the frequencies of other C57 and 129 alleles should be similar in F2 mice grouped by Hbb haplotype. Barring the influence of co-inherited genes, therefore, the observed association between intraerythrocyte glutathione distribution and Hbb haplotype can be confidently attributed to allelic variation in the mouse beta globin gene complex. Based on these results we conclude that allelic variation in mouse Hbb genetically determines the erythrocyte concentration of reactive beta globin sulfhydryl groups, which in turn influences the distribution of glutathione between free and hemoglobin-bound compartments.
This conclusion has important implications for the study of mouse biology and the use of some mouse strains as models for the study of human biology. Mouse Hbb appears to be unique among mammalian beta globin genes in that distinct haplotypes (e.g., HbbS, HbbD, and HbbP) are maintained in homozygous and heterozygous combinations in wild mouse populations (1). The existence of multiple prevalent Hbb haplotypes in wild mice suggests that homozygosity and heterozygosity have adaptive and evolutionarily selective advantages. Our results suggest that adaptive advantage is likely a function of beta globin cysteine sulfhydryl group concentration. If so, then allelic variation in mouse Hbb was likely selected based on the interactions of mouse beta globins with glutathione and NO (or even zinc, which binds to and stabilizes sulfur groups; Ref. 25).
The integrated activities of hemoglobin, glutathione, and NO influence how organisms use oxygen and nitrogen. For example, S-glutathionyl hemoglobin has a higher oxygen affinity (9), whereas S-nitrosohemoglobin transports NO used in vasoregulation (8, 26). GSH is a substrate in enzymatic detoxification of xenobiotics and reactive oxygen species (27–29), whereas NO and oxygen radicals produced by macrophages are essential for host defense (30). It seems likely that beta globin cysteine sulfhydryl group concentration could influence the availability of GSH for enzymatic detoxification reactions (e.g., glutathione peroxidase degradation of hydrogen peroxide to water, or glyoxalase conversion of glyoxal and methylglyoxal to glycolate and
Why mice developed this apparently unique beta globin chemistry is not certain, but some speculation is warranted. Virtually all common human hemoglobin polymorphisms (e.g., those responsible for sickle cell disease, HbE disease, or thalassemia) evolved as adaptations to malaria parasites (Plasmodium sp), a dominant evolutionary pressure in tropical environments (31). Significantly, inbred mouse strains are notoriously variable in their susceptibility to malaria and other pathogens. For example, C57 (HbbS) mice are more resistant to malarial infection than 129 or BALB/c mice (both HbbD) when survival was used as an indicator of susceptibility (31). C57 mice also are more resistant to trypanosome infection than BALB/c mice (32). Jaffe et al. (33) compared malaria infection in C57BL/6, A/J (HbbD), and BALB/c mice and showed that C57 mice were the easiest to infect, but also that strain susceptibility to initial infection was inversely related to strain ability to mount an immune response following intravenous immunization with gamma-irradiated sporozoites.
Malaria parasites invade erythrocytes and are highly susceptible to alterations in intracellular redox equilibrium (34). The host immune response to infectious disease produces NO and oxygen radicals that are used in host defense processes but also contribute to the pathology of infectious disease. Given the role of beta globin cysteine sulfhydryl groups in glutathione and NO metabolism, it is not difficult to imagine that Hbb haplotype could have a significant impact on how different mouse strains respond to pathogenic infection. Mouse strains identified as resistant or susceptible to malarial infection (31) are not uniquely HbbS or HbbD haplotype, although a preponderance of susceptible strains appear to be HbbD haplotype. Consequently, if allelic variation in Hbb evolved as an adaptive response to parasitic infection, it likely did so through a modifying effect of Hbb alleles on other genes involved in host defense.
The results of our experiments suggest that there are two primary reasons why Hbb haplotype should be carefully considered when selecting mouse strains for use as models in the study of human biology. One is the reasonable expectation that mice with beta globins that behave most like human beta globin would be better models of human biochemistry and physiology. Unlike in mice, essentially all human beta globins (with two rare exceptions, Hb Rainier [Tyr145Cys; Ref. 35] and Hb Porto Alegre [Ser9Cys; Ref. 36]) contain only one reactive cysteine residue (β Cys93). Unlike in wild mouse populations, Hbb homozygosity is the norm in human populations. These facts strongly suggest that inbred or hybrid mice with the homozygous HbbS haplotype and monocysteinyl beta globins are inherently more appropriate models than HbbD or HbbP mice, especially when studying processes influenced by systemic redox status.
A second reason to consider Hbb haplotype in strain selection is the likelihood that Hbb is a modifier gene in mice but not in humans. Modifier genes are polymorphic genes that affect the phenotypic expression of other genes (37, 38). They are largely responsible for genetic background effects (i.e., phenotypic differences observed in different mouse strains caused by genomic influences on the expression of specific alleles). A role for mouse Hbb as a modifier gene was proposed by Leder et al. (39), who showed that homozygous HbbS/S mice, genetically modified to express a gene that caused alpha thalassemia, had less severe disease symptoms than similarly modified homozygous HbbD/D mice. Wild mice are born with one of three possible reactive beta globin sulfhydryl group concentrations: 1.0, 1.5, or 2.0 moles of β Cys13 + β Cys93 per mole of beta globin. Because of the apparent effect of Hbb haplotype on glutathione metabolism, the presence of polycysteinyl beta globins can be expected to modify the expression of a host of genes regulating systems that involve glutathione and NO. The developmental implications of genetically determined baseline differences in erythrocyte redox status are legion. These results strongly reinforce the importance of rigorous control of genetic background in the production of genetically modified mice (40). They also suggest that Hbb genotype (or phenotype by cIEF or other means; Refs. 4, 41) should be routinely used as a critical marker of genomic uniformity in genetically modified mice.
These results show that genetic variation in mouse beta globin cysteine content strongly influences intraerythrocyte glutathione metabolism. Due to the influence of polycysteinyl beta globins on systemic redox status, we conclude that homozygous or heterozygous HbbD mice with poly-cysteinyl beta globins are unsuitable and inappropriate for use as models for the study of many aspects of human biology. Progress in biomedical research is highly dependent on the use of animal models, especially mice, given their cost, suitability for genetic manipulation, and widespread use in molecular biology. Of mice and men, all mice are not created equal, and a mouse is not a human. Our results show, however, that some mouse beta globins are chemically and phenotypically more human than others, and suggest this fact should be carefully considered in the selection of mouse strains as models for humans.
Hemoglobin Variants and Intraerythrocyte Glutathione Distribution in Inbred and Hybrid Mice with Different Hbb Haplotypes a

Amino acid sequences of mouse HbbS (β S) and HbbD (β Dmajor and β Dminor) beta globins. Vertical lines signify sequence homology, whereas asterisks identify sites where amino acid sequences vary. Arrows mark the locations of β Cys13 and β Cys93.
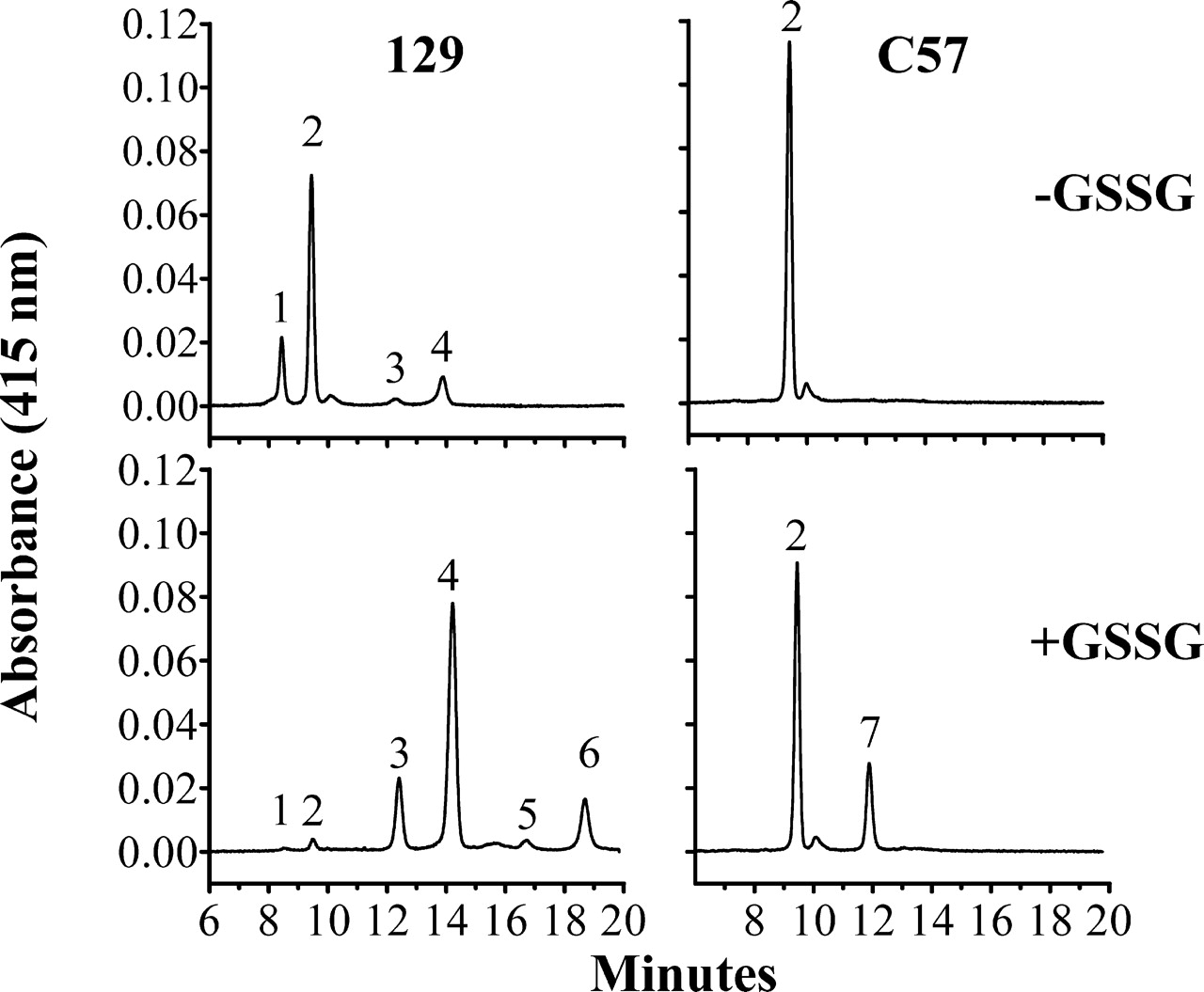
Capillary isoelectric focusing of structural and post-translationally modified hemoglobin variants in inbred C57 and 129 mice. Hemolysates from 129 and C57 mice were incubated with ( + GSSG) or without (−GSSG) 5 mM GSSG for 5 hrs at 37° C. Peak identification and pIs: (1) α β Dminor, 7.20; (2) α β S or α β Dmajor, 7.10; (3) S-glutathionyl-β Cys13 α β Dminor, 6.88; (4) S-glutathionyl-β Cys13 α β Dmajor, 6.78; (5) di-S-glutathionyl α β Dminor; (6) di-S-glutathionyl α β Dmajor; (7) S-glutathionyl-β Cys93 α β S.


Capillary isoelectric focusing of structural and post-translationally modified hemoglobin variants in F2 hybrid mice with different Hbb haplotypes. Hemolysates were obtained from F2 mice with homozygous HbbS/S, heterozygous HbbS/D, or homozygous HbbD/D haplotypes. Peak identification is the same as in Figure 2.
Footnotes
The Research Institute for Children funded these studies.
Acknowledgements
We thank Fred Rambeau for his early contributions to mouse studies that led to this experiment, and Saika Somjee for her help developing the glutathione assay.