
Abstract
The role of gap junctions formed by connexins (Cxs) has been implicated in the homeostatic regulation of multicellular systems. Primitive hematopoietic progenitor cells form a multicellular system, but a previous report states that Cx32 is not expressed in the bone marrow. Thus, a question arises as to why Cx molecules are not detected in the hematopoietic tissue other than in stromal cells. Based on our preliminary study, which suggested a potential impairment of hematopoiesis in Cx32-knockout (KO) mice, the objectives of the present study were to determine whether Cx32 functions in the bone marrow during steady-state hematopoiesis and to examine its possible protective roles during regeneration after chemical abrasions and during leukemogenesis after the administration of a secondary genotoxic chemical, methyl nitrosourea (MNU). As a result, the Cx32 molecule, functioning in the hematopoietic stem cell (HSC) compartment during steady-state hematopoiesis, was observed for the first time; the expressions of Cx32 at the mRNA level, as determined by polymerase chain reaction analysis, and at the protein level, determined using an anti-Cx32 antibody, were observed only in the lin−c-kit+ HSC fraction, using a combination of immunobead-density gradient and immunomagnetic bead separation. Hematopoiesis was impaired in the absence of Cx32, and it was delayed during regeneration after chemical abrasion with 5-fluorouracil at 150 mg/kg body wt in Cx32-KO mice. Cx32-KO mice showed increased leukemogenicity compared with wild-type mice after MNU injection; furthermore, in a competitive assay for leukemogenicity in mice that had been lethally irradiated and repopulated with a mixed population of bone marrow cells from Cx32-KO mice and wild-type mice, the resulting leukemias originated predominantly from Cx32-KO bone marrow cells. In summary, the role of Cx32 in hematopoiesis was not previously recognized, and Cx32 was expressed only in HSCs and their progenitor cells. The results indicate that Cx32 in wild-type mice protects HSCs from chemical abrasion and leukemogenic impacts.
Introduction
Connexin (Cx) functions in the organization of cell-cell communication via gap junctions in multicellular organisms. Gap junctions have been implicated in the homeostatic regulation of various cellular functions, including growth control and differentiation (1), apoptosis (2), and the synchronization of electrotonic and metabolic functions (3).
Radiation exposure and acute tissue injury induce the disconnection of Cxs, resulting in tissue damage (4). On the other hand, the disconnection of Cxs during acute-phase cellular injury also seems to be a protective response that results in active tissue proliferation and consequent recovery. However, transgenic mice expressing a dominant-negative mutant of Cx32 show a notably delayed recovery after partial hepatectomy compared with wild-type mice (5), which implies that the downmodulation of Cx32 is not always advantageous for tissue recovery, despite the finding that a lack of gap junctional restriction seems to enhance cell proliferation (6) (see also Ref. 7 for current information).
Gap junctions are downmodulated during an acute exposure to promoter chemicals, the carcinogenic relevance of which is as yet not clearly understood (8). Temme et al. found that not only spontaneous hepatic tumors but also diethyl-nitrosamine–induced tumors are preferentially induced in Cx32-knockout (KO) mice compared with wild-type mice (9). Why does the downmodulation of Cxs attenuate the protection from malignancy? The reason is that the downmodulation of Cxs results in individual potentially transformable initiated cells that are undergoing independent and infinite growth without interference from surrounding cells; thus, the downmodulation of Cxs in this case seems unlikely to play a protective role (6). On the other hand, the downmodulation of Cxs after exposure to a possible carcinogenic chemical, cadmium, induces cells to undergo apoptosis, which appears to be a protective role (10), though not all cells undergo apoptosis, unfortunately.
The role of Cxs in hematopoietic organs is poorly understood, except in that the expression of Cx43 between hematopoietic progenitor cells and bone marrow stromal cells sustains hematopoiesis (11–14). As Cxs are essential molecules for multicellular organisms, Cxs that organize cell-cell communication within the hematopoietic progenitor cell compartment are surmised to be present in the bone marrow tissue. Recently, we have observed a functional impairment of the bone marrow in Cx32-KO mice in our benzene exposure experiment (15). Krenacs and Rosendaal previously reported that Cx32 is not expressed in the bone marrow (16). If Cx32 is expressed, such Cx32-expressing cells are likely to be rare; for instance, solely in hematopoietic stem/progenitor cells. Hence, similarly to the case of transforming growth factor-β expression, which is observed only in an immature progenitor cell compartment of the bone marrow (17, 18), it seems to be worth studying the expression of Cx32 in the hematopoietic system, particularly in hematopoietic stem/progenitor cells. In this study, we determined whether Cx32 functions solely in primitive hematopoietic cells in a steady-state bone marrow to elucidate its potential protective role during regeneration after bone marrow abrasion and during leukemogenesis after the administration of a secondary genotoxic chemical, methylnitrosourea (MNU).
Cx32-KO mice, first established in 1997 by Nelles et al., can be used for the analysis of the function of Cx32 using a reverse biologic approach (19). In using these mice, the contribution of Cx32, not only in steady-state hematopoiesis and regenerating hematopoiesis but also in the prevention/suppression of leukemogenesis, was elucidated.
Materials and Methods
Experimental Animals.
Cx32-KO mice (Cx32−/− or Cx32−/Y) were genetically modified from the F1 embryonic cell line, 129/J, and the C57BL/6 strain by K. Willecke (19), who kindly provided these Cx32-KO mice, which were backcrossed with the C57BL/6 strain, and maintained as heterozygous mice (Cx32−/+) at the animal facility of the National Institute of Health Sciences (NIHS), Japan. Because the Cx32 gene is X chromosome linked, male mice carrying the homozygous knockout genotype (Cx32−/Y) were generated by mating heterozygous females (Cx32−/+) with wild-type males (Cx32+/Y). The pups were genotyped by polymerase chain reaction (PCR) screening of DNA obtained from their tails.
Eight-week-old C57BL/6 female mice from Japan SLC (Hamamatsu, Japan) were used as the recipients of bone marrow transplantation. All experimental protocols involving laboratory mice in this study were reviewed by an externally established peer review panel, the Committee of the Ethics of the Research and Welfare of the Experimental Animals of the NIHS, and thereby approved by the Animal Care and Use Committee at the NIHS with the experimental code 224-37009639415-2002. Approved experiments were humanely performed in strict accordance with Guidelines for the Care and Use of Laboratory Animals, NIHS, Japan.
Blood and Bone Marrow Separation.
Peripheral blood was collected from the orbital sinus. The numbers of peripheral white blood cells, platelets, and red blood cells were measured using a Coulter counter (Sysmex K-4500; Sysmex Co., Kobe, Japan). Bone marrow cells were harvested from the femur of each mouse (20) after animals were sacrificed by cervical dislocation under deep anesthesia with ethyl ether. A 26-gauge needle was inserted into the femoral bone cavity through the proximal and distal ends of the bone shafts, and bone marrow cells were flushed out under pressure by injecting 2 ml α-minimum essential medium (MEM) with ribonucleosides and deoxyribonucleosides (Invitrogen Corp., Carlsbad, CA). A single-cell suspension was obtained by gently and repeatedly drawing bone marrow cells through a 26-gauge needle and then a 27-gauge needle.
Antibodies.
For immunobead-density gradient separation, the biotinylated antibody cocktail (BD Biosciences, San Jose, CA) containing anti-mouse CD3e (145–2C11), CD11b (M1/70), CD45R/B220 (RA3-6B2), Ly-6G and Ly-6C/Gr-1 (RB6-8C5), and TER-119/erythroid cell (TER-119) antibodies; and the monoclonal antibody cocktail SpinSep (StemCell Technologies Inc., Vancouver, BC, Canada) containing anti-CD5/Ly-1, CD45R, CD11b/Mac-1, Ly-6G/Gr-1, TER119, and 7/4/neutrophil antibodies were used as lineage (lin) markers. As a secondary antibody for the former biotinylated antibody cocktail, streptavidin–peridinin chlorophyll, a protein (PerCP; BD Biosciences) was used. For the latter cocktail, SpinSep, an optimized combination antibody cocktail against SpinSep that had been coated on dense microparticles, SpinSep Mouse Dense Particles (StemCell Technologies Inc.), was used for immunoprecipitation.
For immunomagnetic bead separation, CD117/c-kit conjugated with phycoerythrin (PE; StemCell Technologies Inc.) was used as a progenitor marker, and an anti-PE tetrameric antibody complex (StemCell Technologies Inc.) was used as secondary antibody.
For flow cytometric analyses, the same antibody cocktails from BD Biosciences were used as lineage markers. In addition, a mouse anti-Cx32 monoclonal antibody from two sources (Chemicon International Inc., Temecula, CA, and Santa Cruz Technology Inc., Santa Cruz, CA) was used as the primary antibody for Cx32. As a secondary antibody, anti-mouse Ig conjugated with fluorescein isothiocyanate (FITC; BD Biosciences) was used.
For immunohistochemical analysis, the same anti-Cx32 antibody (Chemicon International, Inc.) was used as the primary antibody. As the secondary antibody, a biotinylated horse anti-mouse IgG antibody (Vector Laboratories Inc., Burlingame, CA) was used, and streptavidin labeled with peroxidase and 3,3′-diamino-benzidine (DAB) was used to detect immunoreactivity (Vector Laboratories Inc.).
Enrichment of Bone Marrow Cells in lin−c-kit+ Fraction.
The lin−c-kit+ fraction is rich in hematopoietic stem cells (HSCs). To obtain a large number of lin−c-kit+-enriched fraction in the bone marrow cells, preseparation was carried out by the combination of immunobead density gradient and immunomagnetic bead separation. First, for the depletion of lineage-positive bone marrow cells, harvested bone marrow cells were processed through an immunobead density gradient using a density-matched medium and dense microparticles coated with a cocktail of an optimized combination of antibodies, SpinSep. Second, for selection of the c-kit+ fraction, immunomagnetic bead separation using magnetic nanoparticles with a magnetic holder was carried out using the manufacturer’s instruction (StemCell Technologies Inc.). For each procedure, the antibodies used are described in the subsection Antibodies in Materials and Methods.
Flow Cytometric Analysis Using Anti-Cx32 Antibody.
Bone marrow cells with or without fractionation for lin−c-kit+ HSC enrichment were stained with the biotinylated antibody cocktail for streptavidin-PerCP, c-kit–PE, the anti-Cx32 antibody, and anti-mouse IgG conjugated with FITC. For exposure to the intracytoplasmic epitope of the anti-Cx32 antibody, cells were fixed with paraformaldehyde and then permeabilized with phosphate-buffered saline supplemented with HEPES and saponin (21). Flow cytometric analysis was carried out using FACS Vantage (BD Biosciences).
Irradiation.
In the assay of hematopoietic progenitor cells, as well as in the repopulation bioassay for leukemogenesis (22), recipient mice were exposed to a lethal radiation dose of 915 cGy at a dose rate of 124 cGy/min using a 137Cs-gamma irradiator (Gammacell 40 Exactor; MDS Nordin Inc., Ottawa, ON, Canada) with a 0.5-mm aluminum-copper filter.
Assay for Colony-Forming Units in Spleen (CFU-S).
The Till and McCulloch method was used to determine the number of hematopoietic spleen colonies (CFU-Ss) (23) formed by hematopoietic progenitor cells. Aliquots of a bone marrow cell suspension were used for evaluating the number of CFU-Ss. Spleens were harvested 9 days after the bone marrow transplantation to determine the number of CFU-S-9 and 13 days to determine the number of CFU-S-13, and then were fixed in Bouin solution. Macroscopic spleen colonies were counted under an inverted microscope at magnification ×5.6. It was previously shown using the C57BL/6 strain that all colonies visible on Day 9 and Day 13 originate from the transplanted bone marrow cells under the condition that the recipient mice were exposed to a lethal radiation dose of 915 cGy (24).
Assay for Granulocyte-Macrophage Colony-Forming Units (CFU-GMs).
CFU-GMs were assayed in semisolid methylcellulose culture (20, 24). Briefly, 8 × 104 bone marrow cells suspended in 100 μl α-MEM were added to 3.9 ml culture medium containing 1% methyl-cellulose (Nakarai-Tesque Co. Ltd., Kyoto, Japan), 30% fetal calf serum (HyClone Laboratories Inc., Logan, UT), 1% bovine serum albumin (Sigma, St. Louis, MO), 10−4 M mercaptoethanol (Sigma), and 10 ng/ml murine granulocyte macrophage colony-stimulating factor (GM-CSF; R&D Systems Inc., Minneapolis, MN). One-milliliter aliquots containing 2×104 cells were placed in 35-mm tissue culture wells (Nalgen Nunc International, Rochester, NY) in triplicate, and were incubated for 6 days in a fully humidified incubator at 37°C with 5% CO2 in air. Colonies were counted using an inverted microscope at magnification ×40 (Olympus Optical Co. Ltd., Tokyo, Japan).
PCR Analysis for Genotyping.
To detect Cx32 wild-type and Cx32-KO alleles, PCR analysis was performed using genomic DNA extracted from the tail of each mouse or from the hematopoietic tissues, spleen and bone marrow, or from tumor cells of the mice in the carcinogenesis tests, and synthetic oligonucleotides were used as primers (19). Hepatic tissues were assayed as the positive control materials (19). To detect the wild-type allele, the common 5′ primer (ccataagtcaggtgtaaaggagc) and the 3′ primer (agataagctgcagggaccatagg) were used; to detect the Cx32-KO allele, the common 5′ primer and neo-primer (atcatgcgaaacgatcctcatcc) were used.
Reverse Transcription (RT) and PCR Analysis of Cx32 Expression.
The expression of the gene encoding Cx32 was analyzed by RT followed by PCR. The total RNA from the bone marrow cells and other tissues was isolated using a Qiagen RNAeasy kit (Qiagen, Valencia, CA). Since hepatocytes are known to express Cx32 (19), the liver was used not only as the hematopoietic organ, but also as the positive control in the verification by RT-PCR analysis. RT was performed using total RNA with random hexamers as primers, according to the instructions provided with the RT kit from Applied Biosystems (Foster City, CA). PCR amplification was performed using the following previously designed oligonucleotide primers including β-actin primers, an amplification control for RT-PCR: Cx32-RT5, 5′-atgcacgtagcctcaccaacagcac-3′; Cx32-RT3, 5′-actcgtagccagcgagaaaagtcg-3′; murine β-actin-5′, 5′-gtaccacgggcattgtgatg-3′; and murine β-actin-3′, 5′-cgttctatcgtgtcgaagag-3′ (15).
Single-Dose Administration of MNU.
Mice were randomly assigned to groups and individually housed. Immediately before use, MNU (Nakarai-Tesque Co. Ltd.) was dissolved in citrate buffer (0.01 M sodium citrate and 0.14 M NaCl, pH 5.5) and injected ip into the mice (25, 26).
Leukemogenicity Bioassay.
Leukemogenicity was determined by a conventional whole-body bioassay and a transplantation bioassay (22). In the conventional whole-body assay, twelve 8-week-old Cx32-KO male mice (Cx32−/Y) and ten wild-type littermates (Cx32+/Y) were injected ip with MNU at 50 mg/kg body wt. In the transplantation bioassay, aliquots of single-cell suspension of the bone marrow (1 × 106 cells) from 8-week-old Cx32−/Y or Cx32+/Y male mice were injected into the tail vein of 8-week-old, 915-cGy–irradiated, wild-type female recipient mice. Only male mice were used as donors and only female mice were used as recipients to utilize the Y chromosome–specific sequence (a candidate testis-determining gene) for differentiating donor-derived neoplasms from recipient-derived neoplasms (27, 28). To study the effect of competitive repopulation on leukemogenicity, a group of mice was also injected with a mixture of cells, one half of which were Cx32-KO bone marrow cells and the other half wild-type bone marrow cells (mixture group). In this procedure, the numbers of CFU-S-9 transferred into each recipient mouse were 3.2 (wild type), 3.1 (mixture group), and 2.6 (Cx32-KO) × 102. In this transplantation bioassay, bone marrow cells from Cx32-KO or wild-type mice were equally effective in protecting against the lethal dose of radiation, and bone marrow cellularity nearly reached that of the steady state after 4 weeks (data not shown). Four weeks after transplantation, 36 and 45 recipient mice were injected ip with MNU at 50 and 75 mg/kg body wt, respectively. The mice were supplied with water ad libitum. The mice in both the conventional leukemogenicity whole-body bioassay and in the transplantation bioassay were monitored throughout their lifetime at least twice daily. Those showing symptoms of advanced leukemia, such as anemia and palpable splenomegaly, were euthanized at the agonal period and then examined hematopathologically. Additionally, mice that died were subjected to gross and microscopic examinations (26).
Histopathological Examination.
For the evaluation of hematopoietic malignancies caused by the injection of MNU in wild-type and Cx32-KO mice, mice from each group were sacrificed under ethyl ether anesthesia for necropsy. For the histopathological examination, all the visceral organs, including the thymus, spleen, sternum, and femoral bone marrow, were fixed in 4% neutral-buffered formalin for 24 hrs. The sternum and femoral bone marrow were decalcified in 7.5% formic acid for 72 hrs. After routine processing, paraffin-embedded sections were stained with hematoxylin and eosin and then examined histopathologically using a light microscope (22).
Immunohistochemical Staining.
To confirm the cellular location of Cx32-positive progenitor cells, spleen colonies were examined by immunohistochemical staining with the anti-Cx32 antibody. Spleen sections containing colonies were fixed with 4% paraformaldehyde solution and embedded in paraffin for thin sectioning. The thin sections were then immunohistochemically stained with the anti-Cx32 antibody, a biotinylated secondary antibody, a horse anti-mouse IgG antibody, and streptavidin labeled with peroxidase to form the ABC complex with 3,3′-DAB.
Statistical Analyses.
The data obtained were stored in a computer and processed for statistical analyses using the Kaplan-Meier method for survival curves and the log-rank test for their statistical significance. The Student t-test was used to evaluate the significance of differences in blood cell count, bone marrow cellularity, and the numbers of progenitor cells, CFU-GMs, CFU-S-9s, and CFU-S-13s between the wild-type group and the KO group. The incidence of hematopoietic neoplasms was evaluated by Fischer exact test. Differences with a P value <0.05 were considered significant.
Results
Expression of Cx32 in Hematopoietic Progenitor Cells and Its Function in Steady-State Hema-topoiesis.
Expression of Cx32 in Hematopoietic Cells.
Figure 1A shows the expression of Cx32 in various lympho-hematopoietic tissues of wild-type mice. As previously reported (19), Cx32 was detected at the mRNA level only in the hepatic tissue by RT-PCR analysis, but was not detected in the spleen, bone marrow, and thymus.
Expression of Cx32 in Hematopoietic Spleen Colonies Developed from Progenitor Cells.
We next studied Cx32 expression in colonies developed in the spleen of lethally irradiated wild-type recipient mice following injection of bone marrow cells from wild-type mice or Cx32-KO donor mice. Hematopoietic spleen colonies are rich in immature cells rather than in cells from peripheral blood or unfractionated bone marrow (24). As shown in Figure 1B, the expression of Cx32 detected by RT-PCR analysis was observed only in the hematopoietic spleen colonies but derived from wild-type bone marrow cells (a1 through a3), not in the colonies derived from Cx32-KO bone marrow cells (c1 and c2). Cx32 expression also was detected in the spleen colonies in Cx32-KO recipient mice repopulated with wild-type bone marrow cells (b1 and b2).
Immunohistochemical staining with the anti-Cx32 antibody was carried out to examine the hematopoietic spleen colonies originating from bone marrow cells from wild-type mice and Cx32-KO mice (Fig. 2). A colony originating from a wild-type bone marrow cell (Fig. 2Aa) shows mild and mottled staining in beige, whereas a colony originating from Cx32-KO bone marrow cells are negative in staining (Fig. 2Ba and b). Interestingly, in a colony observed at a higher magnification (Fig. 2Ab), cells from wild-type mice positively stained by the anti-Cx32 antibody were only scattered in the outer boundary of spleen colonies (circled by dotted line in Fig. 2Aa and arrows in Fig. 2Ab), indicating that the incidence of primitive progenitor cells was still low in the spleen colonies.
Expression of Cx32 in Hematopoietic Stem Cell Compartment.
We then next determined whether Cx32-positive cells are consistently found in the HSC compartment. First, the lin−c-kit+ HSC-enriched fraction was obtained by the combination of immunobead-density gradient separation for depleting lineage-positive cells and immunomagnetic bead separation for selecting c-kit+ cells, followed by flow cytometric analysis using the anti-Cx32 antibody. As a result, the separated lin−c-kit+ HSC fraction was 0.25% with respect to the original unfractionated wild-type bone marrow cells. Figure 3 shows the flow cytometric distribution of the lin−c-kit+ HSC-enriched fraction (Fig. 3B) compared with original unfractionated cells (Fig. 3A) from both wild-type bone marrow cells (the horizontal axis for lineage markers and the vertical axis for c-kit). In Figure 3B, the percentage of lin−c-kit+ compartment (HSC compartment) indicated by an asterisk is 90.2% of the lin−c-kit+ HSC-enriched preseparated fraction. Furthermore, the number for lin−c-kit+ compartment (asterisk in Fig. 3B) is 106.9 times enriched compared to the fraction of the original unfractionated bone marrow cells, as shown in the enclosed corresponding square (Fig. 3A). To determine which fraction Cx32-positive cells belong to, bone marrow cells from wild-type mice and Cx32-KO mice were stained with lineage-PerCP, c-kit–PE, and Cx32-FITC with or without the lin−c-kit+HSC enrichment. In Figure 3C, 28.8% of the lin−c-kit+fraction of wild-type bone marrow cells was found to be Cx32 positive (unshaded profile) compared with the same fraction of bone marrow cells obtained from Cx32-KO mice (shaded profile), which was used as the negative control. Together with frequency data of the lin−c-kit+HSC-enriched fraction, Cx32-positive cells are calculated nearly 0.27% with respect to the original unfractionated whole bone marrow cells.
Whether the mature cell fraction, a lin+c-kit− fraction, contains Cx32-positive cells, the fraction of wild-type bone marrow cells (unshaded profile) is compared to that of the control profile from Cx32-KO mice (shaded profile), as shown in Figure 3D. Since both fractions are nearly identical, few cells may be positive for Cx32 in the lin+c-kit− fraction (0.27% of the lin+c-kit− fraction; Fig. 3D). Cx32-positive cells are 0.0093% with respect to the original unfractionated whole–bone marrow cells (data not shown).
Function of Cx32 in the Steady-State Hematopoiesis.
The steady-state hematopoiesis of wild-type mice was compared to that of Cx32-KO mice. Figure 4A shows the comparison of the absolute body weight, splenic weight, and cellularity of the bone marrow. There were essentially no differences in any of these parameters between wild-type mice and Cx32-KO mice. However, the number of white blood cells and that of platelets were significantly different between wild-type mice and Cx32-KO mice, as shown in Figure 4B. Regarding the decrease in the number of white blood cells, there was no trend toward decrease between numbers of lymphocytes and neutrophils. Moreover, there was no difference in the number of red blood cells. Regarding the number of CFU-GMs, there was a significantly lower number of progenitor cells per unit number of bone marrow cells in Cx32-KO mice than in wild-type mice. Hematopoietic progenitor cells that form CFU-S-9s are considered to be more mature than those that form CFU-S-13s (29, 30). As shown in Figure 4C, in terms of the maturation stages from CFU-S-13 and CFU-S-9 to CFU-GM, the number of all of the hematopoietic progenitor cell compartments of Cx32-KO mice was lower than that of the wild-type mice. Therefore, the present study clearly showed that Cx32 deficiency induced an impaired hematopoiesis specifically in the immature progenitor cell fraction, and changes in differentiated cells may be a reflection of those in immature cells. Thus, Cx32 is assumed to be required for the maintenance of immature hematopoietic progenitor cells.
Function of Cx32 During Growth of Hematopoietic Progenitor Cells.
Although the stromal cell–dependent connexin Cx43 is known to function in cultured stromal cells (12, 13), few cells were positive in anti-Cx32 antibody in the bone marrow. Thus, one question is to answer is whether the hematopoietic defect observed in Cx32-KO mice exists solely in hematopoietic progenitor cells, or also in stromal cells. Accordingly, to examine whether Cx32 deficiency has a negative effect on hematopoiesis in stromal cells of Cx32-KO mice, lethally irradiated wild-type mice and Cx32-KO mice were repopulated with bone marrow cells from wild-type mice or Cx32-KO mice. Results from four different combinations are shown in Figure 5. Regardless of the expression of Cx32 in stromal cells in either Cx32-KO recipient mice or wild-type recipient mice, there were no statistically significant differences in the number of spleen colonies (CFU-S-9s) between the pair of groups that received either wild-type bone marrow cells (Fig. 5A, two left columns) or Cx32-KO bone marrow cells (Fig. 5A, two right columns). Thus, Cx32 deficiency in progenitor cells is concluded as a major factor that is responsible for the production of a significantly small number of colonies. As observed in Figure 4C, it is confirmed that the number of colonies is larger when donor bone marrow cells are from wild-type mice than when they are from Cx32-KO mice.
In Figure 5B, the size of spleen colonies in each group is shown. Significantly smaller colonies were observed in the three groups in which recipient mice, donor bone marrow cells, or both were from Cx32-KO mice rather than in the group in which both recipient mice and donor bone marrow cells were from wild-type mice (Fig. 5B, open column). Because there was no significant difference in size between groups repopulated with Cx32-KO bone marrow cells, the major factor for producing small colonies (Fig. 5B, two right columns) also is assumed to be responsible for Cx32 deficiency in donor progenitor cells, rather than any factor from stromal cells. Concerning the group that received wild-type bone marrow cells (Fig. 5B, the second shaded column from the left), Cx32-KO mice produced significantly smaller colonies due to as yet undetermined reasons.
Regeneration Potency of Bone Marrow Cells from Cx32-KO Mice and Wild-Type Mice.
Treatment with 5-fluorouracil (5-FU) induces a temporary arrest of hematopoietic progenitor cell proliferation, except in the very immature hematopoietic progenitor cell compartment (30–32), in which mature hematopoietic progenitor cells are killed, whereas immature dormant HSCs selectively survive. The number of CFU-GMs per unit number of bone marrow cells was counted for 2 weeks after the 5-FU treatment. As shown in Figure 6, the number of CFU-GMs in both wild-type mice and Cx32-KO mice increased rapidly after 5-FU treatment; however, the increase in the number of CFU-GMs seemed to be delayed in Cx32-KO mice compared with that of wild-type mice.
Experimental Leukemogenesis: Whole-Body Bioassay and the Transplantation Bioassay With or Without Cx32.
Cx32-KO Hematopoietic Progenitor Cells and Leukemogenesis.
A high incidence of hematopoietic neoplasms was observed during the MNU-induced leukemogenesis in Cx32-KO mice. The survival curves of the mice of each group showed that the MNU-treated mice, regardless of genotype, died much earlier because of MNU-induced hematopoietic malignancies and other diseases (Fig. 7A). Untreated control mice, regardless of genotype, gradually started to die 400 days after the treated groups received an MNU.
The percentage of incidences of hematopoietic malignancies in mice treated with MNU, both wild-type and Cx32-KO, and mice in both nontreated control groups are shown in Figure 7B. When Cx32 was knocked out, the incidence of hematopoietic malignancies started to increase rapidly 100 days after MNU treatment (closed squares), which exceeded the incidence of hematopoietic malignancies in wild-type mice (50.0%), and reached 91.7% (P < 0.05 by Fischer exact test). The incidence of hematopoietic neoplasms in nontreated groups of both wild-type and Cx32-KO mice as reference groups increased gradually, reaching 40.0% for the wild-type mice and 33.3% for Cx32-KO mice. Concomitantly, hepatomas developed in both groups of mice after 664 days of age, and the incidence in Cx32-KO mice was higher than that in wild-type mice, although it was statistically less significant (33.3% and 10.0%, respectively; data not shown).
Assay of Leukemogenicity: The Transplantation Assay in Cx32-KO Bone Marrow Cells and Wild-Type Bone Marrow Cells.
Because the incidence of hematopoietic malignancies was significantly high in Cx32-KO mice (Fig. 7B), but the development of hematopoietic malignancies was interfered with by malignancies from other tissues and organs due to competitive risk of the tumorigenicity (data not shown and Ref. 22), lethally irradiated same wild-type mice were repopulated with either bone marrow cells from wild-type mice or Cx32-KO mice, and the development of hematopoietic neoplasms after a single dose of MNU was observed under the same recipient conditions (the transplantation assay). However, few differences in survival time and incidence of neoplasms were observed between mice repopulated with wild-type bone marrow cells and those repopulated with Cx32-KO bone marrow cells (data not shown).
Competitive Assay of Leukemogenicity Between Cx32-KO Bone Marrow Cells and Wild-Type Bone Marrow Cells.
A mixed population of bone marrow cells from Cx32-KO and wild-type mice was injected into lethally irradiated wild-type mice, and the incidence of hematopoietic malignancies competitively caused by bone marrow cells from Cx32-KO mice and those from wild-type mice was determined under the same in vivo conditions of the recipient (the competitive assay). Figure 8 shows the incidences of hematopoietic malignancies in mice that received a single dose of MNU at either 75 mg/kg body wt (dark squares) or 50 mg/kg body wt (medium squares), compared with the nontreated control (light squares), which are plotted against the days after MNU treatment. The incidence of hematopoietic malignancies of the 75 mg/kg body wt MNU-treated group reached 88.9%, whereas that of the 50 mg/kg body wt MNU-treated group reached 33.3%. The incidences of hematopoietic neoplasms that were observed in the competitive assays are shown in Table 1. In the mice treated with MNU at 50 mg/kg body wt and in those treated with 75 mg/kg body wt, two and eight hematopoietic neoplasms developed, respectively.
Samples from these neoplasms were analyzed for their genotype to determine whether the neoplasms originated from bone marrow cells of wild-type mice or Cx32-KO mice (Fig. 9). In Figure 9, lanes 1, 2, 4, 5, 6, 8, and 9 show the presence of the gene inserted for the knockout strategy; thus, the neoplastic samples in these lanes were identified as having originated from bone marrow cells of Cx32-KO mice. The origins of the hematopoietic neoplasms are shown in Table 1. The results show that the malignancies originated from the bone marrow cells of Cx32-KO mice in two of two leukemias in the 50 mg/kg body wt MNU-treated group, and in seven of eight leukemias in the 75 mg/kg body wt MNU-treated group.
Discussion
The role of Cx32 in steady-state hematopoiesis and its potential protective role of prevention during leukemogenesis were analyzed in this study. In this study we demonstrated for the first time that a Cx gene, namely, the Cx32 gene, is expressed in hematopoietic stem/progenitor cells, and in the case of Cx32-KO mice, the regeneration of bone marrow after chemical abrasion was clearly delayed, which suggests a beneficial role of Cx32 in the regeneration. Furthermore, the incidence of MNU-induced leukemia was clearly high in Cx32-KO mice after a single administration of MNU, as shown not only in the whole-body assay (Fig. 7) but also in the competitive repopulation assay using bone marrow cells from Cx32-KO and wild-type mice (Figs. 8 and 9). These results are compatible with the observation of epithelial tumorigenesis in the liver and lungs observed in the same Cx32-KO strain (9), and this is the first observation in leukemogenesis.
Various Cxs are expressed in stromal cells of the fetal liver (Cxs 43, 45, 30.3, 31, and 31.1) and bone marrow (Cxs 43, 45, and 31; Ref. 13). However, the contribution of Cxs to hematopoiesis was found only on the basis of the effect of Cxs via stromal cell dependence; consequently, no Cxs were previously found in hematopoietic stem cells and/or progenitor cells (16). For us this is interesting, because hematopoietic progenitor cells possess morphologic evidence as well as functional evidence for cellular communication with each other (33, 34). Interestingly, in our recent study, Cx32-KO mice exposed to benzene showed a hematopoietic impairment; however, the site of this impairment was not identified in either hematopoietic progenitor or stromal cells (15).
Thus, we first determined whether hematopoietic progenitor cells express Cx32 molecules; however, as reported elsewhere, Cx32 is not detected in the bone marrow (Fig. 1A and Refs. 15 and 19). Interestingly, hematopoietic spleen colonies derived from hematopoietic progenitor cells were found to express Cx32 (Fig. 1B). This observation was further supported by the immunohistochemical reaction of cells in the colonies to the anti-Cx32 antibody, in which Cx32-positive cells were only scattered along the border of each colony (Figs. 2Aa and b). Furthermore, flow cytometry using the anti-Cx32 antibody after performance of the combination of immunobead-density gradient separation and the immunomagnetic bead separation showed that the Cx32-positive fraction was found to belong to the HSC compartment and was calculated as only 0.27% with respect to the unseparated bone marrow cells (Fig. 3). These findings may be in good agreement with a previous report of the absence of Cx32 expression in the bone marrow tissue (13). A hematopoietic disadvantage in progenitor cells associated with Cx32 deficiency was further evident, because all progenitor cells from the bone marrow of Cx32-KO mice showed a ~20% decrease in numbers of CFU-S-13s, CFU-S-9s, and CFU-GMs. Thus, it can be concluded that Cx32 is required for maintaining normal hematopoiesis, specifically during the maturation of hematopoietic stem cells to the progenitor cells.
However, whether Cx32 also is functional in differentiated mature blood cells is questionable, despite the observation that the numbers of white blood cells and platelets were significantly lower in the peripheral blood of the Cx32-KO mice than in the wild-type mice (Fig. 4B). It is of interest to calculate a probability of Cx32-positive cells based on this ratio of those Cx32-positive bone marrow cells out of the lin+c-kit− fraction; that is, only 0.0093% with respect to that of unfractionated original bone marrow cells (data not shown). Because our repeated analyses failed to detect Cx32 expression in mature blood cells, the decreased numbers of white blood cells and platelets in the Cx32-KO mice are regarded as a reflection of the shortage of immature progenitor cell compartments due to the lack of Cx32 at the level of stem cells and progenitor cells.
The bone marrow transplantation in different combinations of the donor and recipient, which was repopulated with bone marrow cells from either wild-type mice or Cx32-KO mice, showed a small number of spleen colonies in the groups repopulated with Cx32-KO bone marrow cells. Interestingly, as shown in Figure 5B, colonies derived from the same Cx32-KO bone marrow cells showed significantly smaller colonies regardless of the genotype of recipients—that is, wild-type or Cx32-KO mice—presumably owing to the lack of Cx32 expression in the hematopoietic progenitor cells (shaded column second from the right vs. closed column far right). The reason why a small size of colonies observed in the Cx32-KO recipient mice received wild-type bone marrow cells cannot be answered in the present study. It is possible that Cx32 deficiency in combination with a lethal dose of whole-body irradiation for the bone marrow transplantation induces an unknown synergistic damage. Our previous observation that Cx32-KO mice treated repeatedly with a dose of benzene by inhalation showed a severe chemical-induced persistent pulmonary injury (15) may be relevant to the present observation. Stem cell regeneration after chemical abrasion with 5-FU was delayed in Cx32-KO mice (Fig. 6), which indicates that early recovery of mice also requires the growth of hematopoietic progenitor cells expressing Cx32. This is compatible with the observation of transgenic mice expressing a dominant-negative mutant of Cx32, which showed a notably delayed recovery after partial hepatectomy (5).
The role of Cx32 is associated with the prevention of carcinogenicity, as an initiation of leukemogenicity was preferentially induced in Cx32-KO mice by a single dose of MNU; thus, Cxs likely have a protective function against leukemogenicity, specifically for the initiation of the carcinogenic process. Phenotypically, the results are compatible with the observation that spontaneous hepatic tumors and diethyl-nitrosamine–induced hepatic tumors tended to develop in Cx32-KO mice compared with wild-type mice (9). Furthermore, radiation-induced hepatocarcinogenesis and diethyl-nitrosamine–induced pulmonary tumorigenesis showed a high frequency of tumorigenesis in Cx32-KO mice (35, 36), which also is compatible with the results of the present study.
Why does the lack of Cxs result in more frequent carcinogenesis? Why was the incidence of leukemogenesis higher in Cx32-KO mice (Fig. 7B, closed squares)? Furthermore, why did leukemogenicity in the wild-type mice appear earlier than that in Cx32-KO mice, although the total incidence remains lower by about 50% (Fig. 7B, open squares) than in the Cx32-KO mice (Fig. 7B, open squares vs. closed squares, respectively). The present study implies that Cx32-KO mice showed a high frequency of leukemogenesis due in part, to a possible suppression of apoptosis of hematopoietic progenitor cells after exposure to chemical carcinogens, and thereby the initiation of leukemogenicity was induced frequently in Cx32-KO mice. Cx32 is, therefore, surmised to protect hematopoietic progenitor cells from leukemogenic triggers in the wild-type mice.
The present competitive assay clearly showed that Cx32-KO bone marrow cells have a higher risk of becoming leukemogenic. The above-mentioned findings in this study imply that Cxs play an essential role in tumor suppression, although a temporary disconnection of Cxs induced by so-called carcinogenic promoter chemicals might induce an independent growth of possible neoplastic candidates, which may, however, eventually undergo apoptosis or be enclosed by cells with recovered Cx function.
Lastly, our results indicate that the risk of developing leukemia in patients with X chromosome–linked Cx32 deficiency, called Charcot-Marie-Tooth syndrome, might not be incidental.
Transplantation Bioassays for Repopulation with Mixture of Bone Marrow Cells Followed by Induction of Tumor by MNU, and Genotyping of Tumor Origin
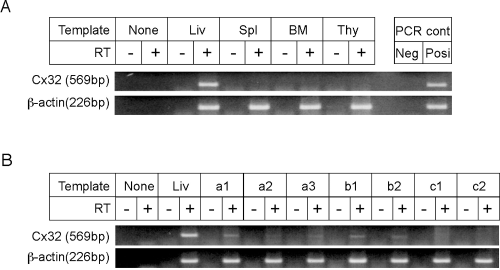
Expression of Cx32 in the lympho-hematopoietic tissues and hematopoietic spleen colonies. (A) Expression of Cx32 in lympho-hematopoietic tissues. Total RNAs were extracted for RT-PCR from the liver (Liv), spleen (Spl), bone marrow (BM), and thymus (Thy) of wild-type mice. Note that Cx32 expression was not detected in the spleen, bone marrow, or thymus, but was detected in the liver, a positive control (see Materials and Methods). PCR: for “Neg” lane and “Posi” lane, no template and whole genome extracted from the tail were loaded, respectively. RT(+) and RT(−): With or without Avian reverse transcriptase, 2.5 U/20 μl, respectively (see Materials and Methods). (B) Expression of Cx32 in hematopoietic spleen colonies (see Materials and Methods). Lethally irradiated wild-type mice were injected with bone marrow cells from wild-type or Cx32-KO donor mice. After 9 days, total RNAs extracted from individual hematopoietic spleen colonies derived from wild-type bone marrow cells or those from Cx32-KO bone marrow cells were reverse transcribed followed by PCR and then loaded (a1–a3, c1 and c2). Total RNAs extracted from colonies derived from wild-type bone marrow cells removed from the lethally irradiated Cx32-KO recipient mice followed by repopulation with wild-type bone marrow cells were similarly loaded (b1 and b2).

Cells in spleen colonies immunohistochemically stained with anti-Cx32 antibody. (A) Spleen colonies derived from wild-type bone marrow cells (a and b). (B) Cx32-KO bone marrow cells (i.e., control for negative staining; subpanels a and b). As shown in Aa and Ba, cells from spleen colonies were found positively stained with a mottled pattern in the former figure and negatively stained in the latter figure. The positively stained cells are located only in the outer boundary of the colony. Dotted line in Aa indicates border of the colony. Ab and Bb show a higher magnification of spleen colonies derived from wild-type mice and Cx32-KO mice, respectively. As shown in Ab, a colony was a mottled pattern with positively stained cells (arrows) in beige. Bb shows the negative control. Spleens were stained with the anti-Cx32 antibody and with the biotinylated secondary antibody, horse anti-mouse IgG antibody, and streptavidin labeled with peroxidase. Bars indicate 200 μm in Aa and Ba, and 25 μm in Ab and Bb.
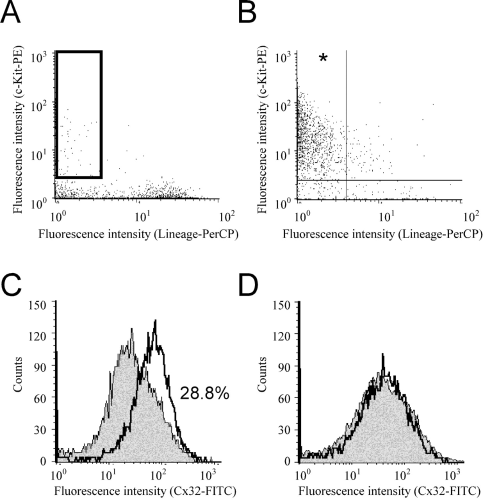
Flow cytometric analyses for lin−c-kit+ HSC-enriched fraction and lin−c-kit+ Cx32-positive cells from wild-type mice. Flow cytometry after bone marrow cell separation carried out by a combination of the immunobead-density gradient separation and the immunomagnetic bead separation. (A) Unseparated bone marrow cells. (B) Bone marrow cells fractionated by combination of the immunobead-density gradient separation for eliminating lineage marker positive cells and the immunomagnetic bead separation for c-kit+cells. The vertical axis in both figures indicates fluorescence intensity for the PE-labeled anti–c-kit antibody, and the horizontal axis indicates fluorescence intensity for Per-CP–labeled streptavidin for biotinylated lineage antibodies. The vertical and horizontal lines in panel B indicate the negative and positive borders of fluorescence intensity. The asterisk in panel B indicates the targeted lin−c-kit+compartment and HSC compartment. Note that the corresponding area of the asterisk in panel B is indicated by the square box in panel A. (C and D) Histogram of the FITC-labeled anti-Cx32 antibody. The lin−c-kit+fraction (C) and the lin+c-kit−fraction (D) for wild-type bone marrow cells (open profile with bold line) and the same fraction for Cx32-KO bone marrow cells (shaded profile), a negative control. Cx32-positive fraction in panel C calculated for the lin−c-kit+ fraction in wild-type bone marrow cells is 28.8%.
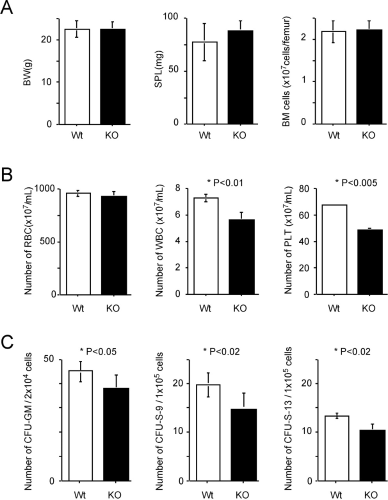
Parameters associated with steady-state hematopoiesis. Row A: from left, body weight (BW), splenic weight (SPL), and bone marrow cellularity (BM cells; n = 6 in each genotype). Row B: from left, numbers of peripheral blood cells—red blood cells (RBCs), white blood cells (WBCs), and platelets (PLTs; n = 6 for each genotype). Row C: from left; numbers of hematopoietic progenitor cells in steady-state CFU-GMs, hematopoietic progenitor cells for CFU-S-9s, and those for CFU-S-13s. Three donor mice were used for each genotype, and six mice were used for each recipient group. Open box, wild type; solid box, Cx32-KO; vertical bar, standard deviation of the mean. *Difference between wild-type and Cx32-KO mice is significant (P values are indicated in each figure).
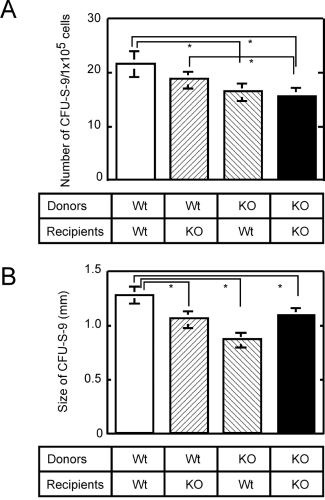
Number and size of CFU-Ss assayed in four different combinations between donors, either wild-type or Cx32-KO bone marrow cells, and lethally irradiated recipients, either wild-type or Cx32-KO mice. (A) Number of CFU-S-9s. (B) Size of CFU-S-9s. *Differences between each bar connected with a line are significant (P < 0.05); three donor mice were used for each genotype, and six mice were used for each recipient group.
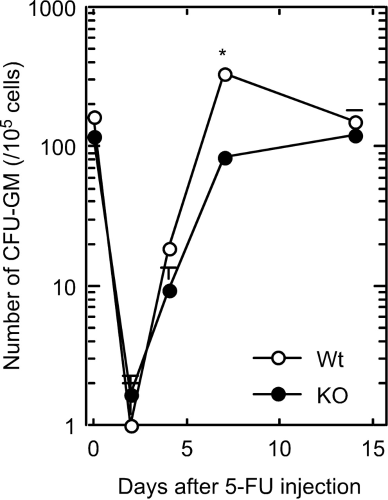
Changes in number of CFU-GMs in femoral bone marrow cells in wild-type or Cx32-KO mice (days after a single dose of 5-FU, iv, 150 mg/kg body wt). Open circle, wild type; closed circle, Cx32-KO. *Seven days after 5-FU injection, at which time the difference between the wild-type and Cx32-KO mice was significant (P < 0.05); three mice each were used for each data point).
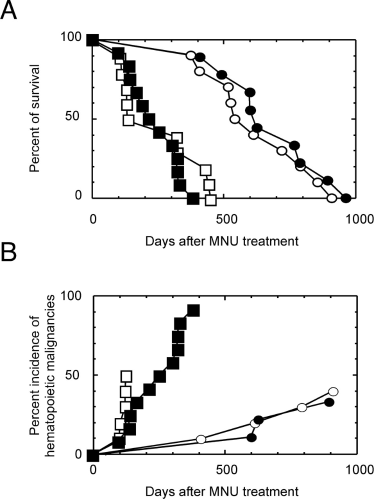
Whole-body assay of hematopoietic malignancies by a single dose of MNU at 50 mg/kg body wt. (A) Survival fraction. (B) Cumulative incidences of hematopoietic malignancies in Cx32-KO mice or wild-type mice with or without a single ip injection of MNU. Open circle, wild type without MNU injection, 10 mice; open square, wild type with MNU injection, 10 mice; closed circle, Cx32-KO without MNU injection, 9 mice; closed square, Cx32-KO with MNU injection, 12 mice.
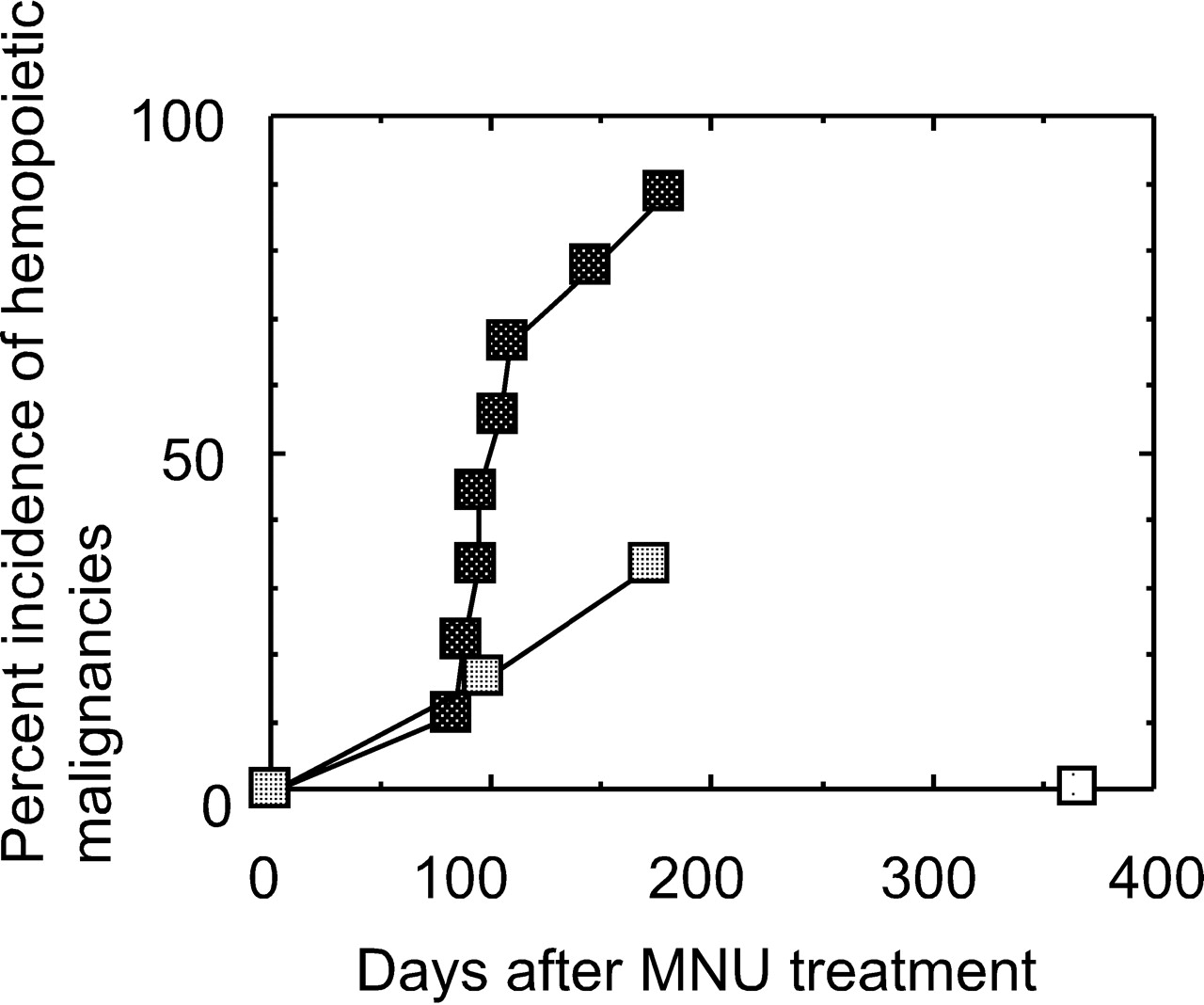
Competitive bone marrow transplantation bioassays repopulated with mixture of bone marrow cells from wild-type and Cx32-KO mice, followed by a single dose of MNU. Cumulative incidences of hematopoietic malignancies in the group repopulated with the mixture cells followed by a single ip injection of MNU at 50 or 75 mg/kg body wt. Light square, vehicle treatment; medium squares, 50 mg treatment; dark squares, 75 mg treatment.

Genotyping of hematopoietic neoplasms whose origin was identified by genomic PCR. N, negative control for PCR without DNA; P, positive control for PCR using genomic DNA from the Cx32+/− hepatic tissue. Lanes 1–9, DNA extracted from hematopoietic neoplasms that developed during assay of bone marrow transplantation. Lane 3, control neoplasm that developed in mouse repopulated with wild-type bone marrow cells. Lane 4, control neoplasm that developed in mouse repopulated with Cx32-KO bone marrow cells. Lanes 1, 2, and 5–9, neoplasms that developed in mice repopulated with a mixture of bone marrow cells from wild-type and Cx32-KO mice. Note that lane 7 has a faint band for the KO allele, which shows an additional simultaneous expression band from the repopulated normal hematopoietic cells. For lanes 1, 2, 5, 6, 8, and 9, the tumors arising from Cx32-KO bone marrow cells show double bands, namely a Wt allele and another allele for KO strategy. Intensities of these bands are identical compared with that of lane 4.
Footnotes
This study was supported by a Grant-in-Aid for Scientific Research B, 11694334, and also by the Japanese Society for the Promotion of Science (JSPS) Invitation Fellowship for Research in Japan, S01275.
1
Current address: Department of Veterinary Medicine, College of Animal Resource, Kangwon National University, Chuncheon, 200-701, Republic of Korea.
Acknowledgements
This study is dedicated to the late Dr. Eugene P. Cronkite for his fifth-year memorial. We thank Dr. Y. Kawasaki, Dr. K. Sai, Ms. E. Tachihara, Ms. N. Moriyama, Ms. Y. Shinzawa, Ms. Y. Usami, Mr. K. Terasaka, and Mr. Morita for excellent technical assistance, and Ms. N. Kikuchi, Ms. M. Yoshizawa, and Ms. M. Hojo for secretarial assistance.