
Abstract
Accumulating evidence links calcium-overload and oxidative stress to atrial remodeling during atrial fibrillation (AF). Furthermore, atrial remodeling appears to increase atrial thrombogeneity, characterized by increased expression of adhesion molecules. The aim of this study was to assess mitochondrial dysfunction and oxidative stress–activated signal transduction (nuclear factor-κB [NF-κB], lectin-like oxidized low-density lipoprotein receptor [LOX-1], intercellular adhesion molecule-1 [ICAM-1], and hemeoxgenase-1 [HO-1]) in atrial tissue during AF. Ex vivo atrial tissue from patients with and without AF and, additionally, rapid pacing of human atrial tissue slices were used to study mitochondrial structure by electron microscopy and mitochondrial respiration. Furthermore, quantitative reverse transcription polymerase chain reaction (RT-PCR), immunoblot analyses, gel-shift assays, and enzyme-linked immunosorbent assay (ELISA) were applied to measure nuclear amounts of NF-κB target gene expression. Using ex vivo atrial tissue samples from patients with AF we demonstrated oxidative stress and impaired mitochondrial structure and respiration, which was accompanied by nuclear accumulation of NF-κB and elevated expression levels of the adhesion molecule ICAM-1 and the oxidative stress-induced markers HO-1 and LOX-1. All these changes were reproduced by rapid pacing for 24 hours of human atrial tissue slices. Furthermore, the blockade of calcium inward current with verapamil effectively prevented both the mitochondrial changes and the activation of NF-κB signaling and target gene expression. The latter appeared also diminished by the antioxidants apocynin and resveratrol (an inhibitor of NF-κB), or the angiotensin II receptor type 1 antagonist, olmesartan. This study demonstrates that calcium inward current via L-type calcium channels contributes to oxidative stress and increased expression of oxidative stress markers and adhesion molecules during cardiac tachyarrhythmia.
Introduction
Cardiac tachyarrhythmias are known to induce significant electrophysiologic and structural changes in cardiac tissue, which themselves may contribute to the persistence and aggravation of atrial fibrillation (AF) (1–8). During AF, there is not only a compromised cardiac function, but also a high risk for thromboembolic events. Altered intracellular calcium homeostasis and angiotensin II receptor type I (AT1R) activation have been identified as important factors contributing to the cellular and cardiac hypertrophy, atrial extracellular matrix (ECM) accumulation, and fibrosis in AF (2–5). Accumulating evidence points to a linkage of oxidative stress and calcium overload to atrial remodeling during AF (for an excellent recent review see Korantzopoulos et al. [9]). Mihm et al. (10) were the first to demonstrate extensive oxidative damage in atrial myocardium from patients with AF, which was mainly mediated by peroxynitrite and was not observed in similar samples from patients in sinus rhythm (SR). Later it was experimentally shown that rapid atrial pacing led to decreased tissue levels of ascorbic acid, whereas protein nitration was increased along with decreased effective refraction period (11). All these changes could be prevented by pretreatment with vitamin C. Several key components of the cardiac excitation-contraction coupling were shown to be highly sensitive to oxidative stress (11–16). Gene expression profiling of atrial tissue samples from patients with SR and AF revealed a decreased expression of antioxidative genes, whereas that of five reactive oxygen species (ROS)-producing genes was increased (16).
Furthermore, AF has substantial effects on mitochondrial structure and function: Lin et al. (17) detected oxidative damage of mitochondrial DNA and demonstrated diminished ATP synthesis along with increased production of ROS that was due to initial calcium overload.
Mainly myocardial NAD(P)H oxidase but also (although to a lesser extent) NO synthase (eNOS) and mitochondrial oxidases are responsible for atrial oxidative stress during AF (18, 19). Angiotensin II (angII), via activation of AT1R, enhances NAD(P)H oxidase-dependent O2 − production (20, 21), a mechanism that, due to the increased expression and activity of the atrial renin-angiotensin system (RAS) during AF (22, 23), results in elevated oxidative stress. Recently, using a rapid pacing model of in vitro differentiated P19 cardiomyocytes, it was demonstrated that tachycardia is associated with mitochondrial swelling and dysfunction, which could be prevented by the blocker of L-type calcium channels, verapamil (24). In a previous study, we demonstrated marked alterations of mitochondrial morphology in the atrial tissue of patients with AF (25). An induction of atrial expression of adhesion molecules that persists even months after successful cardioversion has been observed previously in patients with AF (26, 27).
The purpose of the present study was to assess the effect of AF on mitochondrial dysfunction and oxidative stress–activated signal transduction by analyzing nuclear factor-κB (NF-κB), lectin-like oxidized low-density lipoprotein receptor (LOX-1), intercellular adhesion molecule-1 (ICAM-1), and hemeoxgenase-1 (HO-1). We hypothesized, that during atrial tachyarrhythmia there is an increased oxidative stress, which partly depends on calcium inward current via L-type calcium channels and thereby supports the activation of the redox-sensitive NF-κB signaling pathway and corresponding target gene expression. By comparing results obtained from ex vivo atrial tissue samples from patients with SR or AF, respectively, with data obtained by rapid pacing ex vivo of human atrial tissue slices, we were able to largely exclude the effect of factors other than the elevated frequency itself on mitochondrial function and oxidative stress–induced signaling.
Materials and Methods
Patients.
Right atrial appendages were obtained from 26 patients undergoing cardiac bypass surgery or mitral/aortic valve replacement. Tissue samples were taken from 13 consecutive patients with chronic persistent AF (≥6 months; AF) and from 13 matched patients with SR. Patients were matched for age, history of myocardial infarction, hypertension, and, especially, for their medications, as statins and sartans were expected to have a major impact on the target parameters analyzed in this study. There were differences in the left ventricular ejection fractions and in the presence of valvular diseases. The effect of these differences was assessed by statistical analyses. The clinical characteristics are shown in Table 1.
For in vitro pacing experiments, atrial tissue samples were taken from 50 additional consecutive patients with SR. Patients’ characteristics and the usage of tissue slices in different experiments is specified in Table 2. Tissue slices from one patient are sufficient for one type of analysis only (e.g., for either quantitative reverse transcription polymerase chain reaction [RT-PCR] or immunoblot).
The study conforms with the Declaration of Helsinki and was approved by the local ethic committee. All patients gave written informed consent to participate in the study.
Quantitative RT-PCR.
Samples of human atrial appendages were rapidly frozen in liquid nitrogen and stored until further analysis. Total RNA was prepared as described recently (28) by applying the method of Chomczynski and Sacchi (29). A 1-μg quantity of total RNA was transcribed into cDNA. A 25-μl reaction mixture consisted of 1× SensiMix (Quantace, London, UK), 0.5 μl of SYBR-Green I (Quantace), 1 μl cDNA, and 0.3 μM of the specific primers listed in Table 3. PCR was performed in an iCycler (BioRad, Munich, Germany). Quantities of GAPDH mRNA were used to normalize cDNA contents.
Immunoblot Analysis.
Tissue homogenates and nuclear extracts were prepared as previously described (28). The primary antibodies against human LOX-1/SR-E1 (polyclonal goat IgG, dilution in Tris-buffered saline [TBS] 1:1000, R&D Systems, Wiesbaden, Germany), ICAM-1 (sc-8439 mouse monoclonal IgG2α, 1:500, Santa Cruz, CA), HO-1 (monoclonal mouse IgG1, clone HO-1–1, dilution in TBS 1:1000), GAPDH (MAB374 mouse monoclonal IgG, 1:1000, Chemicon Europe, Hampshire, UK), or Phospho-IκB-α (Ser32/36, mouse monoclonal IgG, 1:1000, Cell Signaling, Frankfurt/M., Germany) together with the secondary anti-goat (1:5000, Dianova, Hamburg, Germany) or anti-mouse (1:2000, Cell Signaling) horse-radish peroxidase–conjugated antibodies, and the Super-Signal West Dura Extended Duration substrate (Pierce, Rockford, IL) were used for detection.
Protein carbonylation was assessed by using the OxyBlot Protein Oxidation Detection kit provided by Chemicon, strictly following the recommended protocol. In each case, 20 μg of protein were used for the initial derivatization reaction.
Electrophoretic Mobility Shift Assay (EMSA), Supershift EMSA, and NF-κB p50 Enzyme-Linked Immunosorbent Assay (ELISA).
Preparation of nuclear extracts, EMSA, and supershift EMSA were performed as described (30).
Nuclear amounts of NF-κB p50 were determined quantitatively by using the TransAM NFκB p50 Transcription Factor Assay Kit provided by Active Motif Europe (Rixensart, Belgium) following the instructions of the supplier. Ten micrograms of nuclear extract were applied for each sample.
Culturing Tissue Slices and Ex Vivo Pacing.
Tissue culture and rapid pacing were performed as described recently (24, 28). Previously, it has been shown that tissue slices are kept viable under the applied conditions for at least 8 days and that slow in vitro pacing (0.6 Hz) does not alter tissue response compared with non-paced tissue samples (24, 28). Similarly, we did not observe any significant differences in the mRNA or protein expression levels between samples paced with either 2 or 4 Hz (28). This has been demonstrated for various important cellular signaling pathways that are representative for typical cellular alterations described in response to tachycardia and include the activation of angiotensin, Erk1/2, and calcineurin/NFAT signaling pathways. This finding holds true also for the parameters analyzed in this study. Thus, non-paced tissue slices served as reasonable controls, and pacing at 2 Hz was used for rapid pacing in experiments, where limited amounts of atrial tissue slices from individual patients prevented the simultaneous application of the full range of pacing rates (0, 0.6, 2, and 4 Hz). Pacing at 2 Hz is also supposed to minimize the formation of radicals at the electrodes in comparison to 4 Hz. Verapamil, 1 or 10 μM verapamil (Isoptin, Abbott, Wiesbaden, Germany); 10 μg/ml olmesartanmedoxomil (Sankyo Pharma, Munich, Germany); 5 μM resveratrol and 100 μg/ml apocynin (Sigma, Taufkirchen, Germany); and 0.5 μM NF-κB inhibitor IV (Merck Biosciences, Schwalbach, Germany) were added to the media throughout the pacing period where indicated.
Measurement of Respiration.
Oxygen uptake of up to three atrial tissue slices was measured at 30°C in a thermostat-controlled and gently stirred chamber equipped with a Clark-type electrode (Paar Physica Oxygraph Respirometer, Bioenergetics and Biomedical Instruments, Innsbruck, Austria). Oxygen consumption of the slices was determined in Dulbecco’s modified Eagle’s medium (Sigma). Endogenous as well as maximal (uncoupled) respiration was analyzed. Maximal respiration was induced by the stepwise addition of 0.5 μM of the uncoupler FCCP. Endogenous respiration is expressed as percentage of maximal respiration. This procedure is independent of the size of the used slices. We did not determine absolute values of respiration since oxygen consumption does not only depend on the weight of the slices, but also on the mechanical stress due to gently stirring.
Electron Microscopy.
For electron microscopy, three independent mitochondria or tissue preparations were used for each experimental condition. After sedimentation at 320 g at 4°C, the pellet of isolated mitochondria was fixed with a mixture of 4% formaldehyde (freshly prepared from paraformaldehyde) and 0.4% glutaraldehyde for 1 hr at 4°C. Tissue samples were fixed with the same mixture for 24 hrs at 4°C. Thereafter, the specimens were rinsed thoroughly with phosphate-buffered saline (pH 7.4), postfixed in 1% osmium tetroxide for 1 hr at 4°C, dehydrated in a graded series of ethanol, en bloc contrasted with 1% uranyl acetate in 70% ethanol, and flat-embedded between two polyethylene foils in Durcupan (Fluka/Sigma, Deisenhofen, Germany). In case of isolated mitochondria, each washing and incubation step was followed by sedimentation at 320 g at 4°C. Ultrathin sections (50–70 nm) were prepared with a Leica Ultracut UCT (Bensheim, Germany), mounted on Formvar-coated slot grids, and examined with a Zeiss transmission electron microscope 900 (Oberkochen, Germany).
Statistical Analyses.
The univariate comparisons of the two groups with (AF) and without (SR) atrial fibrillation were done using the t-test (Satterthwaite’s approximation to compute the degrees of freedom) for metric parameters. To assess the possible impact of mitral stenosis (MS), these calculations were repeated without the three patients with MS in the AF group.
In addition, for each parameter analyzed (LOX-1 protein; LOX-1 mRNA; ICAM-1 protein; ICAM-1 mRNA; HO-1 protein; HO-1 mRNA), we performed analysis of variance (ANOVA) with the two main factor groups (“SR” vs. “AF”) and MI (yes vs. no) or group and MS (yes vs. no), respectively, and the cross-effect between these factors.
Differences in the oxidative phosphorylation were analyzed by the Wilcoxon signed rank test. All values are expressed as the mean ± standard error of the mean (SEM). A P value of < 0.05 was considered to be statistically significant.
The in vitro pacing experiments were also analyzed using ANOVA. The values of the experiments are expressed as mean ± SEM. All statistical decisions were made with a critical probability of α = 5% without α-adjustment. The data should be interpreted in an exploratory manner. Statistical analyses were carried out using SAS software (Version 9.1, SAS Institute, Cary, NC).
Results
Oxidative Stress and Redox-Signaling in Fibrillating Human Atrial Tissue.
Protein-Carbonylation in Fibrillating Human Atrial Tissue.
Among the best characterized oxidative protein modifications is the formation of carbonyl groups (31). By using the OxyBlot system, which involves the covalent addition of 2,4-dinitrophenylhydrazine (DNPH) to any available carbonyl groups and subsequent detection of the dinitrophenol (DNP) moieties using anti-DNP antibodies, we elucidated the degree of oxidative stress in atrial fibrillation. As shown in Fig. 1A and B, there was a significantly increased formation of carbonylated protein (PCO) in atrial tissue of patients with AF in comparison with SR (AF: 206% ± 42% vs. SR: 100% ± 12%, P = 0.024, n = 12).
In addition, we observed significantly lower tissue contents of free thiols in atrial tissue from patients with AF when compared with those of patients with SR (AF: 1.217 ± 0.089 vs. SR: 0.813 ± 0.085 mmol/mg protein, P = 0.003, n = 12) (Fig. 1C).
Activation of the NF-κB Signaling Pathway in Fibrillating Human Atrial Tissue.
To demonstrate a possible activation of the redox-sensitive NF-κB signaling pathway during AF, atrial amounts of nuclear NF-κB were assessed in atrial tissue samples from patients with AF or SR by EMSA and supershift EMSA. As exemplarily shown in Figure 5A, there was an increase in the amounts of nuclear NF-κB in AF when compared with SR. These observations could be confirmed by the quantitative determination of nuclear amounts of NF-κB p50 as performed by means of the TransAM p50 ELISA kit. As shown in Figure 5B, atrial tissue from patients with AF contained significantly more nuclear NF-κB p50 (133.1% ± 8.4%) when compared with SR (100% ± 11.9%, P = 0.041, n = 13).
Expression of LOX-1, ICAM-1, and HO-1 in Fibrillating Human Atrial Tissue.
Gene expression of the NF-κB target genes LOX-1, ICAM-1, and HO-1 were quantified using RT-PCR. Amounts of atrial LOX-1 mRNA were increased in patients with AF compared with SR (177% ± 17% vs. SR: 100% ± 20%, P = 0.0035, n = 13) (Fig. 2A). mRNA levels of ICAM-1 were only slightly elevated in AF (124% ± 12% vs. SR: 100% ± 15%, P = not significant, n = 13) (Fig. 2C). Amounts of HO-1 mRNA were also significantly increased in atrial tissue from patients with AF (203.3% ± 41.3% vs. SR: 100% ± 19.7%, P = 0.004, n = 13) (Fig. 2E). At the protein level, amounts of atrial LOX-1 (153% ± 19%; vs. SR: 100% ± 18%, P = 0.029, n = 13), ICAM-1 (197% ± 28% vs. SR: 100% ± 15%; P < 0.001, n = 13), and HO-1 (198.7% ± 45.4%, P = 0.042, n = 12) were significantly elevated during AF (Fig. 2B, D, and F).
To assess additional effects of MI or MS, respectively, on the differences between the SR and AF groups, ANOVAs were performed for each parameter analyzed (LOX-1 protein; LOX-1 mRNA; ICAM-1 protein; ICAM-1 mRNA; HO-1 protein; HO-1 mRNA) with group (“SR” vs. “AF”) and MI or MS, respectively, as main factors and the cross-effect between each two factors. None of these six ANOVAs yielded P-values near the significance level (P < 0.05) for either MI factor or its interaction. Only HO-1 protein appeared to be affected by the presence of MS. Omission from analysis of the three patients with MS in the AF group, which all showed HO-1 protein levels above the mean, increased the P-value for HO-1 protein to 0.21.
Ex Vivo Atrial Tissue Slices.
To confirm the functional role of calcium-mediated oxidative stress during AF, atrial tissue slices from patients without AF were cultured ex vivo and electrically paced for 24 hrs. Functional integrity and viability of the slices is proven by regular bimonthly assessment of their ability to respond to pacing with increased MAP kinase Erk2 mRNA expression or by propidiumbromide staining, respectively, as described previously (28).
Rapid Pacing Impairs Mitochondrial Morphology and Respiration.
Electron microscopy was performed on either mitochondria isolated from atrial tissue (Fig. 3A–H) or on atrial tissue slices directly (Fig. 3I–K). Non-paced controls were characterized by morphologically intact mitochondria exhibiting normal cristae structure (Fig. 3A, B, and I). In contrast, rapidly paced tissue contained increased numbers of pale and swollen mitochondria, which partly showed cristaeolysis, as well as completely disrupted mitochondria (Fig. 3C, D, and J). Verapamil prevented these pacing-dependent changes of morphology (Fig. 3G, H, and K) without affecting non-paced control (Fig. 3E and F). These changes resembled the alterations seen in patients with AF (25).
Next, we determined endogenous oxygen consumption of the atrial tissue slices as a basic parameter of mitochondrial function. It reflects the activity of oxidative phosphorylation. Endogenous respiration was significantly lower in paced tissue compared with non-paced controls (59.4% ± 10.1% vs. 76.5% ± 4.5% of maximum respiration, P = 0.024, n = 5) (Fig. 3L). The impaired oxidative phosphorylation in mitochondria was attenuated by verapamil (endogenous respiration: 69.3 ± 8.5 pmol O2/min/mg protein with verapamil vs. 59.4 ± 10.1 pmol O2/min/mg protein without verapamil, P = 0.043, n = 5) (Fig. 3L).
Pacing of Atrial Tissue Induces Formation of Carbonylated Protein.
The OxyBlot analysis revealed that rapid pacing for 24 hrs induces a significant increase in the amounts of PCO (4 Hz: 250.4% ± 36.1% vs. 0.6 Hz (100%), P = 0.003, n = 5). Verapamil at 10 μM concentration provoked a clear reduction of this elevated protein carbonyl formation (4 Hz + verapamil: 121.2% ± 22.4% vs. 4 Hz, P = 0.016, n = 5) (Fig. 4A and B).
As observed with ex vivo tissue from patients with AF, we also observed in response to rapid pacing in vivo a reduction of cellular free thiols. As shown in Fig. 4C, pacing at 4 Hz reduced the thiol content to 186.1 ± 31.2 mmol/mg protein compared with pacing at 0.6 Hz (242.4 ± 38.4 mmol/mg protein, P = 0.006, n = 4). Again, this loss of thiols could be partly prevented by 10 μM verapamil (217 ± 43.4 mmol/mg protein, P = not significant, n = 5).
Pacing of Ex Vivo Atrial Tissue Slices Activates NF-κB Signal Transduction.
Gel-shift analysis of pooled tissue slices from five independent pacing experiments revealed the nuclear accumulation of NF-κB after rapid pacing at 2 Hz. Supershift was observed by means of anti-p50 and anti-p65 antibodies, indicative of the classical NF-κB pathway being involved (Fig. 5A).
As with ex vivo tissue samples from patients with AF, we could confirm these findings by the quantitative determination of nuclear amounts of NF-κB p50. Using the TransAM p50 ELISA kit (Active Motif), we show a clear trend towards increased nuclear amounts of NF-κB p50 in response to pacing at 4 Hz when compared with slowly paced tissue (4 Hz: 171% ± 32.2% vs. 0.6 Hz: 100% ± 27.1%, P = 0.06, n = 8) (Fig. 5C). These increased amounts of nuclear p50 could be significantly decreased by 10 μM verapamil (94.7% ± 16.3% of 0.6 Hz, P = 0.0016, n = 8). NF-κB inhibitor IV lead to reduction of nuclear amounts of p50 that did not reach statistical significance (117.2% ± 18.1% of 0.6 Hz, n = 4) (Fig. 5C).
Phosphorylation of IκBα is a critical regulatory step in the activation of NF-κB. It targets IκB-α for polyubiquitination and proteasome-mediated degradation. Release from IκBα unmasks the nuclear localization signal of NF-κB and thus mediates its translocation. Rapid pacing increased atrial amounts of phospho-IκBα substantially (2 Hz: 279% ± 62% vs. non-paced: 100% ± 7%; n = 5, P = 0.019). This activation of IκBα was abolished by the administration of 10 μM verapamil (123% ± 27%; P = 0.049 vs. non-paced, n = 5) (Fig. 5D and E).
Pacing-Induced Expression of NF-κB Target Genes LOX-1, ICAM-1, and HO-1.
Rapid pacing increased amounts of LOX-1 mRNA 2.5-fold compared with non-paced control (249% ± 35% vs. 100% ± 20%, P < 0.001, n = 12). Verapamil dose-dependently reduced this tachycardia-dependent induction of LOX-1 mRNA (1 μM: 209% ± 26% of control, P = not significant; 10 μM: 126% ± 33% of control, P = 0.04) (Fig. 6A). Similar changes could be observed at the protein level, where we included slow-pacing (0.6 Hz) and 4 Hz pacing as further controls. As previously observed for various parameters, there were no differences in the atrial response between either 0 Hz vs. 0.6 Hz or 2 Hz vs. 4 Hz, respectively, for LOX-1 or ICAM-1 protein expression detectable. Amounts of LOX-1 protein were raised about 2.3- or 2.5-fold by rapid pacing at 2 Hz (231% ± 36.2%, n = 10, P = 0.002) or 4 Hz (247% ± 55.5%, P = 0.019, n = 8) when compared with non-paced control. The differences were similar when compared with 0.6 Hz (see Fig. 6B for details). Increased LOX-1 expression could be reduced by verapamil (1 μM: 183% ± 52%, P = not significant; 10 μM: 91% ± 11%, P = 0.035 vs. 2 Hz, P = 0.032 vs. 4 Hz) (Fig. 6B and E).
Rapid pacing led also to a pronounced induction of ICAM-1 expression. At the mRNA level, a 4.3-fold increase was observed compared with non-paced controls (437.5% ± 82% vs. 100% ± 21.4%, P < 0.001, n = 12). Verapamil at 10 μM provoked a partial reduction of increased ICAM-1 mRNA amounts (252.9% ± 65.1%, P = 0.04, n = 14). Similar results were obtained at the protein level, where rapid pacing at either 2 or 4 Hz led to a significant increase of ICAM-1 when compared with 0 or 0.6 Hz, respectively. This increase was partially abolished by verapamil (2 Hz: 225% ± 27% vs. non-paced control: 100% ± 24%; P < 0.001, n = 10; 1 μM verapamil: 159% ± 46%, P = not significant; 10 μM verapamil: 127% ± 12.5%, P = 0.027) (Fig. 6C–E). Comparing 4 Hz with 0.6 Hz yielded similar results (Fig. 6D and E).
The changes in gene expression of LOX-1 and ICAM-1 observed after ex vivo pacing of atrial tissue slices largely resembled those observed in patients with AF (Fig. 2).
Additional experiments were performed to prove the frequency-dependency of LOX-1 and ICAM-1 mRNA expression. Whereas pacing at 0.6 Hz did not change expression levels when compared with non-paced controls, pacing at 4 Hz resulted in elevated LOX-1 and ICAM-1 mRNA levels that were equal to those observed in response to pacing at 2 Hz (Fig. 6C–E).
As shown in Figure 7A and B, pacing at 2 Hz led also to a significant increase in HO-1 protein amounts (2 Hz: 396.9% ± 53.1% vs. 100% ± 6.7%, P < 0.001, n = 12). Verapamil at 10 μM abolished this increase in HO-1 expression (124.8% ± 18.7%, P < 0.001, n = 12).
Inhibition of NF-κB Target Gene Expression.
To further explore which stimuli and signaling pathways contribute to oxidative stress–altered gene and protein expression in tachyarrhythmia, we applied in addition to an L-type calcium channel blocker (verapamil), an inhibitor of NADPH oxidase (apocynin), the antioxidants and inhibitors of NF-κB signaling (resveratrol, NF-κB inhibitor IV), and an AT1R blocker (olmesartan medoxomil).
As shown in Figure 7, verapamil effectively prevented the pacing-dependent increase in both HO-1 (2 Hz: 396.9 ± 53.1 vs. 2 Hz + verapamil: 124.8% ± 18.7%, P < 0.001, n = 12) (Fig. 7 A and B) and LOX-1 protein expression (2 Hz: 230.7 ± 36.2 vs. 2 Hz + verapamil: 91% ± 10.7%, P < 0.001, n = 12) (Fig. 7C).
Olmesartan appeared to be nearly equally effective in preventing the pacing-dependent increase of HO-1 (180.5% ± 48.8%, P = 0.046, n = 3)(Fig. 7B) or LOX-1 expression (94.5% ± 2.4%, P = 0.024, n = 3)(Fig. 7C).
Both apocynin and resveratrol led to partial reduction only of pacing-induced HO-1 and LOX-1 expression, which did not reach statistical significance (Fig. 7B and C).
To further verify that the elevated nuclear amounts of NF-κB p50 observed in AF and in response to in vitro pacing contribute to increased expression of the NF-κB target genes, we studied the effect of NF-κB inhibitor IV on mRNA levels of HO-1, LOX-1, and ICAM-1. As expected, the administration of NF-κB inhibitor IV led to a decrease of nuclear amounts of NF-κB p50 (Fig. 5C), which is paralleled by reduced amounts of the target genes analyzed (compared with 2 Hz pacing in the absence of inhibitor) (Fig. 7D).
Discussion
In the present study, by using ex vivo atrial tissue samples from patients with AF and SR as well as rapid pacing in vitro of human atrial tissue slices, we demonstrate that AF is associated with oxidative stress, mitochondrial dysfunction, and oxidative stress–activated signal transduction. The activation of the redox-sensitive NF-κB signaling pathway and elevated expression of its target genes, LOX-1 and ICAM-1, may contribute to an increased risk for platelet and leukocyte adhesion to the atrial endocardium. All these alterations are—to a large extent—calcium-dependent as could be concluded from the protective effects of verapamil.
Accumulating evidence points to the importance of tachycardia-induced “oxidative stress” within atrial and ventricular myocardium (8, 10, 11, 28). Carnes et al. demonstrated the involvement of oxidative and nitrosive stress in “electrical remodeling” in a rapid atrial pacing model (11); they demonstrated that 48 hrs of rapid pacing reduced tissue ascorbate levels and increased protein nitration. In an earlier study, the same group also showed increased rates of protein carbonylation in fibrillating human tissue (10). These results provide evidence that tachycardia increases the concentration of ROS and reactive nitrogen species (RNS). The nitration and carbonylation of structural proteins impair myocardial energetic and electrophysiologic properties. Carnes et al. (11) proposed that oxidative stress mediates the alterations in atrial electrophysiologic parameters such as shortening of the atrial action potential. In the ventricles, tachycardia-induced oxidative stress may be critical for the induction of apoptosis (8). Recently, we showed that rapid pacing of in vitro–differentiated cardiomyocytes for 24 hrs leads to oxidative stress and mitochondrial dysfunction (24).
Results of this study demonstrate the occurrence of oxidative stress in rapidly paced atrial tissue slices as well as in atrial tissue samples derived from patients with AF. The analysis of the activity of oxidative phosphorylation in atrial tissue slices revealed a pacing-dependent impairment of mitochondrial function. A similar reduction of respiration has been previously observed in in vitro–differentiated cardiomyocytes in response to rapid pacing (24). In this model, diminished cellular adenosine triphosphate (ATP) levels due to impaired ATP synthesis were found in addition to reduced respiration. It seems reasonable to assume that similar mechanisms underlie the impairment of mitochondrial respiration observed in in vitro–differentiated cardiomyocytes and atrial tissue slices, respectively, as studied here, in response to rapid pacing. Inhibition of electron transfer within the respiratory chain by impairment of mitochondrial ATP-production either at the level of adenine nucleotide transport or ATP-synthesis causes increased production of superoxide anion radicals by the respiratory chain (32).
This pacing-dependent impairment of mitochondrial respiration seems to be closely associated with morphologic changes of mitochondria. In support of this view, equal swelling and disturbance of cristae structure has been observed in in vitro–differentiated cardiomyocytes (24) and atrial tissue slices in response to rapid pacing, and also in ex vivo right atrial tissue samples from patients with AF. In all these settings, mitochondrial morphology could be largely preserved by limiting calcium influx via blockage of L-type calcium channels with verapamil. It is known that the mitochondrial Ca2+ transport system translates an elevated cytosolic Ca2+ concentration into increased mitochondrial Ca2+ concentration (33). This mechanism mediates swelling of mitochondria and compromises oxidative phosphorylation as has been demonstrated by others (34, 35). Although no increase in mitochondrial Ca2+ has been found during AF by electron probe microanalysis (36), inhibition of mitochondrial Ca2+ influx by ruthenium red had a protective effect in a rat heart model of ventricular tachycardia (37). Our data suggest that inward calcium flux via L-type calcium channels is an important trigger for mitochondrial dysfunction and oxidative stress within myocytes during tachycardia. The finding that verapamil protects from oxidative stress is supported by a recent study from Mason et al. (38). They showed that mibefradil, which blocks L-type and T-type Ca2+ channels, and verapamil prevent the oxidation of cellular constituents and have cytoprotective effects. Mibefradil was found to be more potent than verapamil at preventing lipid peroxide formation. However, because of significant side effects, mibefradil is not used clinically, and therefore the demonstrated verapamil effect appears to be clinically more relevant.
In addition, ROS may be formed in the heart by mitochondria via uncoupling of oxidative phosphorylation. Previously, we did not find significant changes in uncoupled respiration between the paced and non-paced cardiomyocytes applying the uncoupler FCCP (19). This finding is in accordance with previous data showing that the contribution of mitochondrial oxidases to atrial oxidative stress during AF is low.
The importance of oxidative damage to contractile dysfunction during AF has been shown by Mihm and coworkers (10). The increase in carbonylation of myofibrillar proteins in atrial myocytes from patients with AF resulted in the loss of fibrillar function. In the present study, the OxyBlot kit was used for the quantification of protein oxidation in atrial tissue from patients with AF and in in vitro–paced atrial tissue slices. In both settings, increased formation of carbonylated proteins could be detected, which is in line with the findings of Mihm et al. (10). In parallel to the increase in protein carbonylation, we now demonstrate a pacing-dependent loss of free thiol. This occurred in patients with AF in vivo, but also in response to rapid pacing in vitro. Therefore it is reasonable to assume that the increased frequency per se brings about oxidative stress.
Elevated intracellular calcium levels (39) and ROS (40) are major activators of the immediate early response transcription factor NF-κB. Typical target genes of NF-κB are pro-inflammatory cytokines such as interleukin-8 and TNFα, but also the endothelial adhesion molecules ICAM-1 and VCAM-1, and the lectine-like oxidized low-density lipoprotein (oxLDL) receptor, LOX-1. NF-κB contributes also to the induction of HO-1 expression (41). HO-1 is a redox-sensitive inducible protein that contributes to cytoprotection against oxidative stress under a wide range of unrelated conditions (42). The HO-1 promoter contains responsive elements for NF-κB, AP-1 and 2, and Nrf2. The latter appears to be critically important for HO-1 expression (43).
The LOX-1 promoter, too, contains binding sites for numerous transcription factors, including NF-κB and AP-1 (44, 45). Thus, ROS are supposed to represent important activators of LOX-1 gene expression. Nagase et al. (46) were the first to demonstrate that expression of LOX-1 is induced by O2- and H2O2 in cultured aortic endothelial cells (ECs). Previous reports indicated that LOX-1 expression in vitro was regulated by a variety of stimuli including shear stress (47), transforming growth factor-β1 (48), proinflammatory cytokines (49), angiotensin II (50), and oxLDL itself (51). As shown previously, AF is associated with a strong activation of the atrial RAS, including induction of the angiotensin II–generating angiotensin-converting enzyme (ACE) (22, 23). This represents one key mechanism by which atrial NAD(P)H oxidase is activated (20, 21). In support of this view, we observed in this study inhibitory effects of both the AT1R blocker, olmesartan, and of the inhibitor of NAD(P)H oxidase, apocynin, on the induction of NF-κB target genes. Of importance, activation of LOX-1 may further enhance superoxide production (51). We show here an increased expression of LOX-1 in atrial tissue from patients with AF, which is in full accordance with the presence of oxidative stress in fibrillating atrial tissue. This induction of LOX-1 is not only indicative of, and further aggravates the oxidative stress itself, but is assumed to directly contribute to the pro-inflammatory and prothrombotic state. In support of this view, there was a strong increase in ICAM-1 expression in response to rapid pacing of atrial tissue slices and in ex vivo atrial tissue derived from patients with AF. Furthermore, by studying tissue arrays of a large cohort of patients with AF, it could be demonstrated that VCAM-1 expression is increased in AF, too; a finding that could be confirmed in an in vivo model of acute pacing in the pig (52). It has been demonstrated previously that activation of LOX-1 leads to an up-regulation of adhesion molecule expression (53).
Nuclear accumulation of NF-κB p50 was observed in both ex vivo atrial tissue from patients with AF and in atrial tissue slices rapidly paced in vitro. This finding strongly implies that oxidative stress via activation of NF-κB signaling mediates oxidative stress response/prothrombotic target gene induction (ICAM-1, LOX-1), which may account for the endocardial/endothelial activation followed by an increased risk of atrial thrombus formation during AF. As concluded from the supershift analysis performed in this study, the classical p50/p65 NF-κB components are involved in this setting as could be confirmed by the quantitative analyses of nuclear amounts of NF-κB p50.
Inhibitors of NF-κB activation were able to prevent the nuclear accumulation of NF-κB p50, and both antioxidants and NF-κB inhibitors diminished the expression of LOX-1, ICAM-1, and HO-1. This is indicative of NF-κB being involved in the induction of these genes and is in full accordance with their established characteristic as NF-κB target genes. It should be noted, however, that the expression of most genes is not solely dependent on a single transcription factor. AP-1 for example has been shown to strongly affect the expression of typical NF-κB target genes. Thus, under different experimental settings and pathophysiologic conditions, intensive cross-talk of signaling pathways may cause varying degrees of transcriptional activity of individual target genes.
The finding of increased HO-1 expression in atrial tissue from patients with AF and in response to rapid pacing in vitro indicates the existence of oxidative stress during tachycardia and reflects the tissue’s attempt to build up an adequate anti-oxidative defense. This, however, is not achieved as is obvious from the compromised mitochondrial morphology and the failure to prevent the induction of ICAM-1 and LOX-1. Both oxidants and antioxidants have been shown to induce HO-1 expression (54), which again is indicative of different pathways being involved in the induction of transcription.
We show that antioxidants and inhibitors of NF-κB prevent the pacing-dependent induction of not only ICAM-1 and LOX-1, but also of HO-1. This seems to be in contrast to previous work of others demonstrating the induction of HO-1 in response to caffeic acid phenethyl ester (CAPE) and curcumin, two natural compounds with antioxidative and NF-κB inhibiting activity (55). However, this effect very likely is related to the peculiar chemical structures of these compounds, because in the same study resveratrol reduced HO-1 activity. Furthermore, prolonged exposure to CAPE or curcumin (24 hrs) failed to provoke the induction of HO-1 expression and activity observed after 6 hrs. This finding raises the possibility of different regulatory mechanisms being active during acute or chronic episodes of oxidative stress and might well apply to prolonged episodes of tachycardia. Indeed, data of Stahnke et al. (56) suggest that in the pathogenesis of demyelinating diseases, stress-induced HO-1 initially plays a protective role, while its chronic up-regulation shows adverse effects. The cardioprotective activity of resveratrol involves HO-1 but appears to be independent of NF-κB activation (57).
In summary, data from the present study indicate that tachycardia is associated with mitochondrial dysfunction and oxidative stress followed by the activation of the NF-κB signaling pathway with induction of NF-κB target gene expression in atrial tissue. Multiple tachycardia-associated factors appear to contribute to this response, which all are directly or indirectly linked to oxidative stress. Accordingly, AT1R blockade, inhibition of L-type calcium channels, inhibition of NAD(P)H oxidase, applications of antioxidants, and inhibition of NF-κB activation were all found to abolish or decrease the pacing-dependent changes in the atrial tissue. The facts that equal results (observed in ex vivo atrial tissue from patients with AF and in ex vivo rapidly paced tissue samples from patients with SR), together with the observation that verapamil most potently prevented oxidative stress and associated signaling pathway activation, led us to conclude that the elevated frequency per se and concomitant Ca2+-overload precede and induce mitochondrial dysfunction and oxidative stress in AF. These results may also have clinical implications. Atrial ischemia produces an increase in cellular calcium load and oxidative stress in atrial tissue. Thereby, atrial ischemia provides a specific substrate for AF maintenance (58). Recent experimental and clinical data showed that calcium channel blockers have a specific efficacy to prevent AF in this specific situation (24, 59). Thus, the data from the present study provide more information about the potential pathophysiologic mechanism, explaining why calcium channel blockers are effective and useful to attenuate atrial cellular remodeling especially under conditions of increased cellular calcium load and oxidative stress.
Limitations.
Because of the small number of patients included in the study, the data should be interpreted as exploratory. Future studies employing larger patient groups would allow further subdivision according to clinical characteristics. Specifically, the presence/absence of valvular disease is an interesting issue. Studies of Stanley Nattel’s group (60, 61) have shown that AF with and without valvular heart diseases (VHD) exhibit differences with regard to gene expression. Among the genes differentially expressed in AF-VHD vs. SR-VHD were calcium channel subunits. Of interesting, AF-VHD showed reduced L-type calcium current compared with SR-VHD. In the present study, no impact of valvular disease on the differences between SR and AF in the mRNA expression of LOX-1, ICAM-1, and HO-1 as well as on protein amounts of ICAM-1 and LOX-1 was detected.
Studies have shown quantitative differences between left and right atrial tissue. In general molecular changes appear more pronounced in left atrial samples compared with right ones. The principle regulatory mechanisms, however, are found to be largely comparable. Nevertheless, left atrial tissue samples appear to be substantially influenced by concomitant heart diseases like mitral or aortic valve diseases and left ventricular failure. The effect of these factors is much less in right atrial tissue. Thus, right atrial tissue might reflect even better AF-related changes.
Patient Characteristics for Ex Vivo Analyses of Atrial Tissue from Patients with SR and AF
Characteristics of Patients with SR Included in the In Vitro Pacing Experiments
PCR Primers
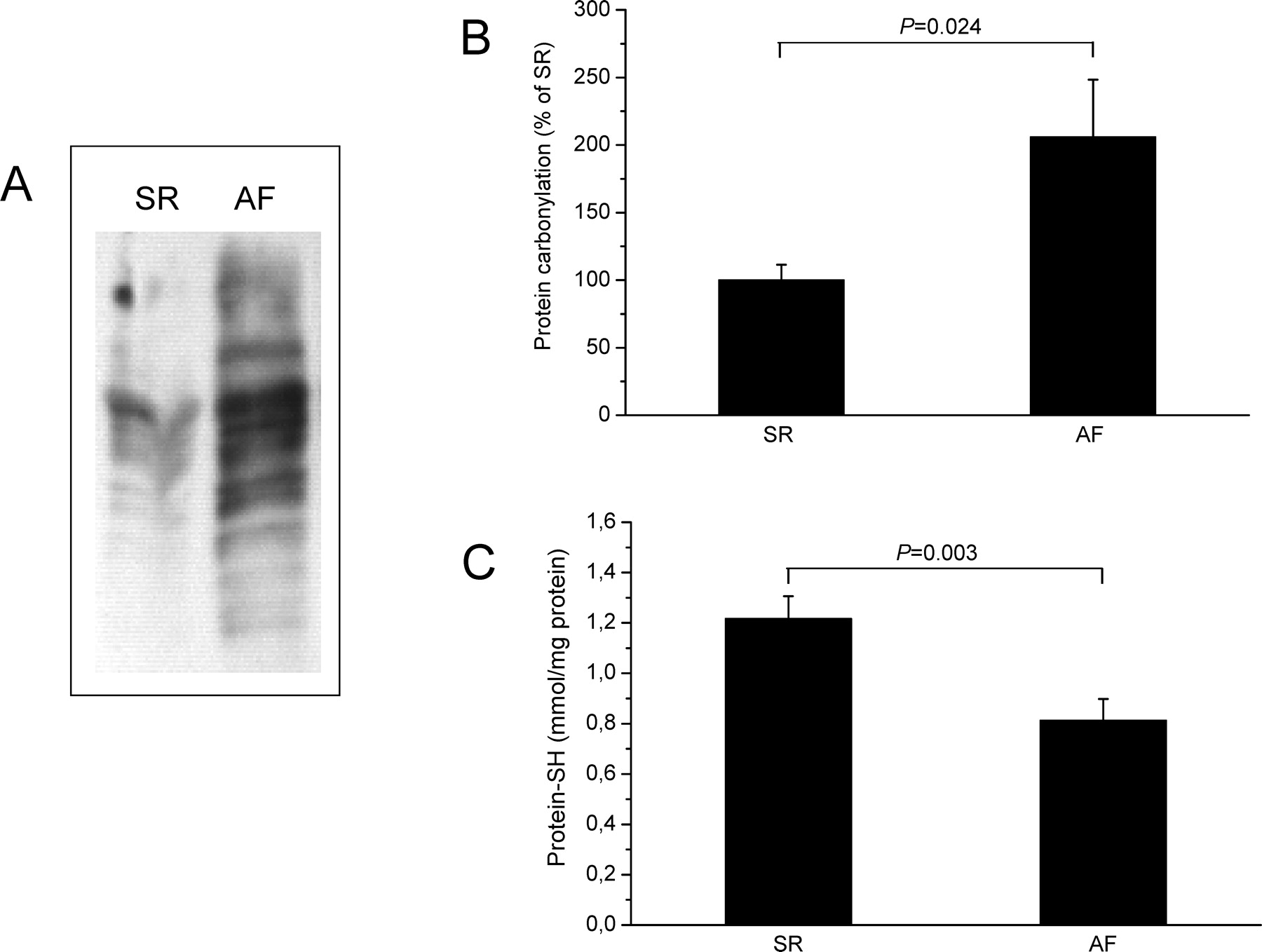
Protein carbonylation (PCO) and protein-SH content in atrial tissue of patients with SR and AF. (A) Typical OxyBlot demonstrating increased PCO formation in fibrillating atria. (B) Quantitative analysis of OxyBlots revealed 2-fold increased PCO formation in atrial tissue from patients with AF (n = 12). (C) Amounts of protein-SH were siginificantly lower in atrial tissue of patients with AF when compared with those of patients with SR (n = 12).

Elevated expression of LOX-1, ICAM-1, and HO-1 in fibrillating human atria. Atrial amounts of LOX-1, ICAM-1, and HO-1 were assessed at the mRNA (A, C, and E) and protein level (B, D, and F) by quantitative real-time reverse transcriptase polymerase chain reaction (RT-PCR) or immunoblot analyses, respectively. In AF, LOX-1 and HO-1 expression was significantly increased during AF (A, B, E, and F). ICAM-1 expression was significantly increased during AF at the protein level (D and E), whereas mRNA levels showed only a tendency towards elevated expression. (G) Representative Western blots demonstrating LOX-1, ICAM-1, HO-1, and GAPDH expression in atrial tissue samples from patients with AF and SR (30 μg of protein/lane) (n = 13).

Effect of rapid pacing on mitochondrial morphology (A–K) and respiration (L). (A–H) isolated mitochondria; (I–L) atrial tissue slices. Electron microscopic appearance of mitochondria of non-paced controls showing preserved morphology and well-defined cristae structures (A, B, and I). Two Hertz pacing of atrial tissue slices induces mitochondrial swelling and partial cristaeolysis (C, D, and J). Verapamil reduces mitochondrial degeneration as fully intact mitochondria remain (G, H, and K) without changing mitochondrial morphology of controls (E and F). Magnification: × 50.000. (L) Oxygen consumption of atrial tissue slices was reduced upon rapid pacing. Verapamil at 10 μM concentration preserved mitochondrial respiration. Mean ± SEM in percentage of untreated controls (n = 5).
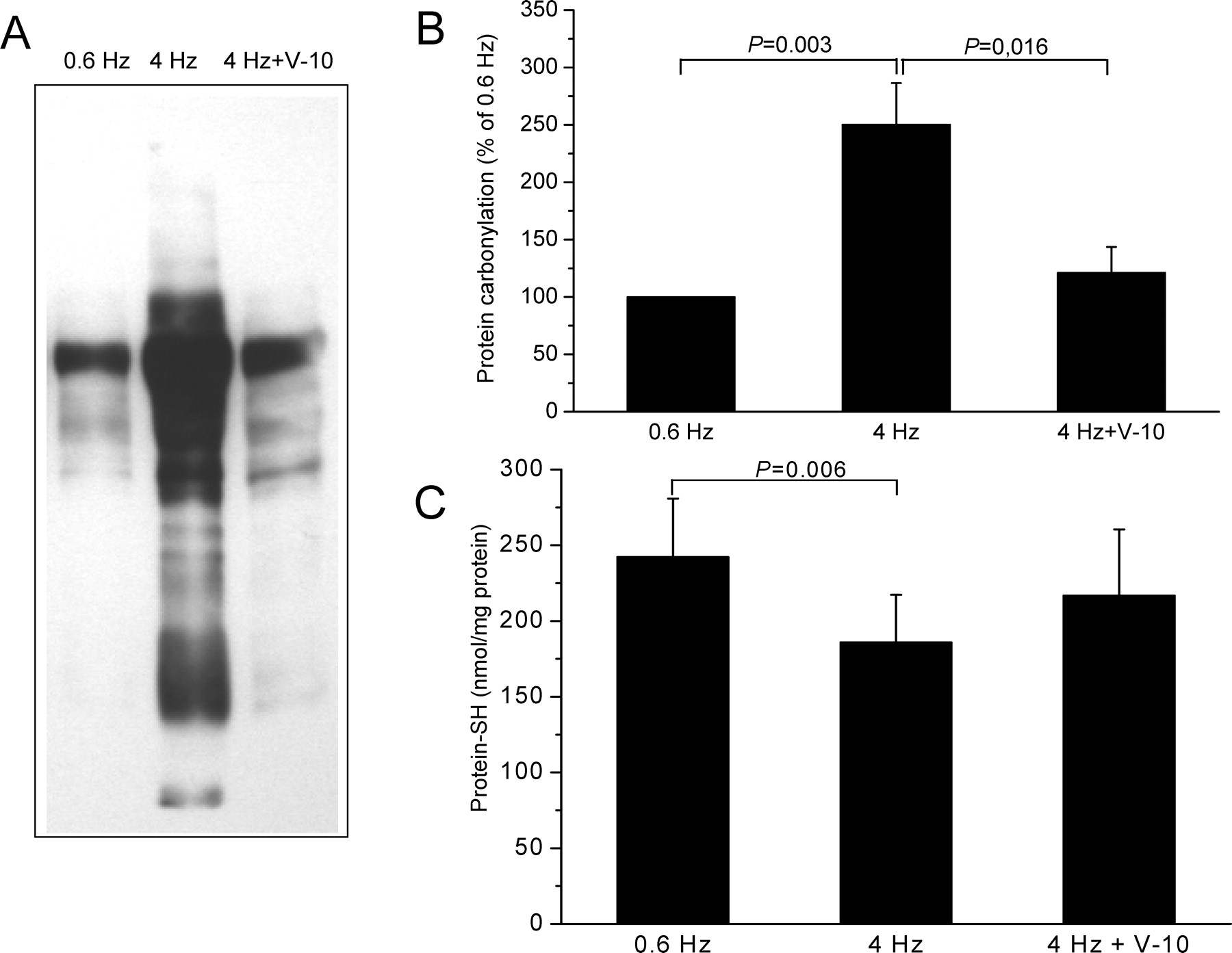
PCO and protein-SH content in atrial tissue slices in response to rapid pacing. (A) Oxyblot showing PCO formation. (B) Quantitative analysis of Oxyblots showing 2-fold increased PCO formation after rapid pacing, which was largely prevented by 10 μM verapamil (PCO levels of 0.6 Hz were 89% of those observed in AF (Fig. 1), n = 5). (C) Amounts of protein-SH were significantly lower in atrial tissue in response to rapid pacing at 4 Hz when compared with pacing at 0.6 Hz. Verapamil at 10 μM partly restored the SH levels (n = 5).
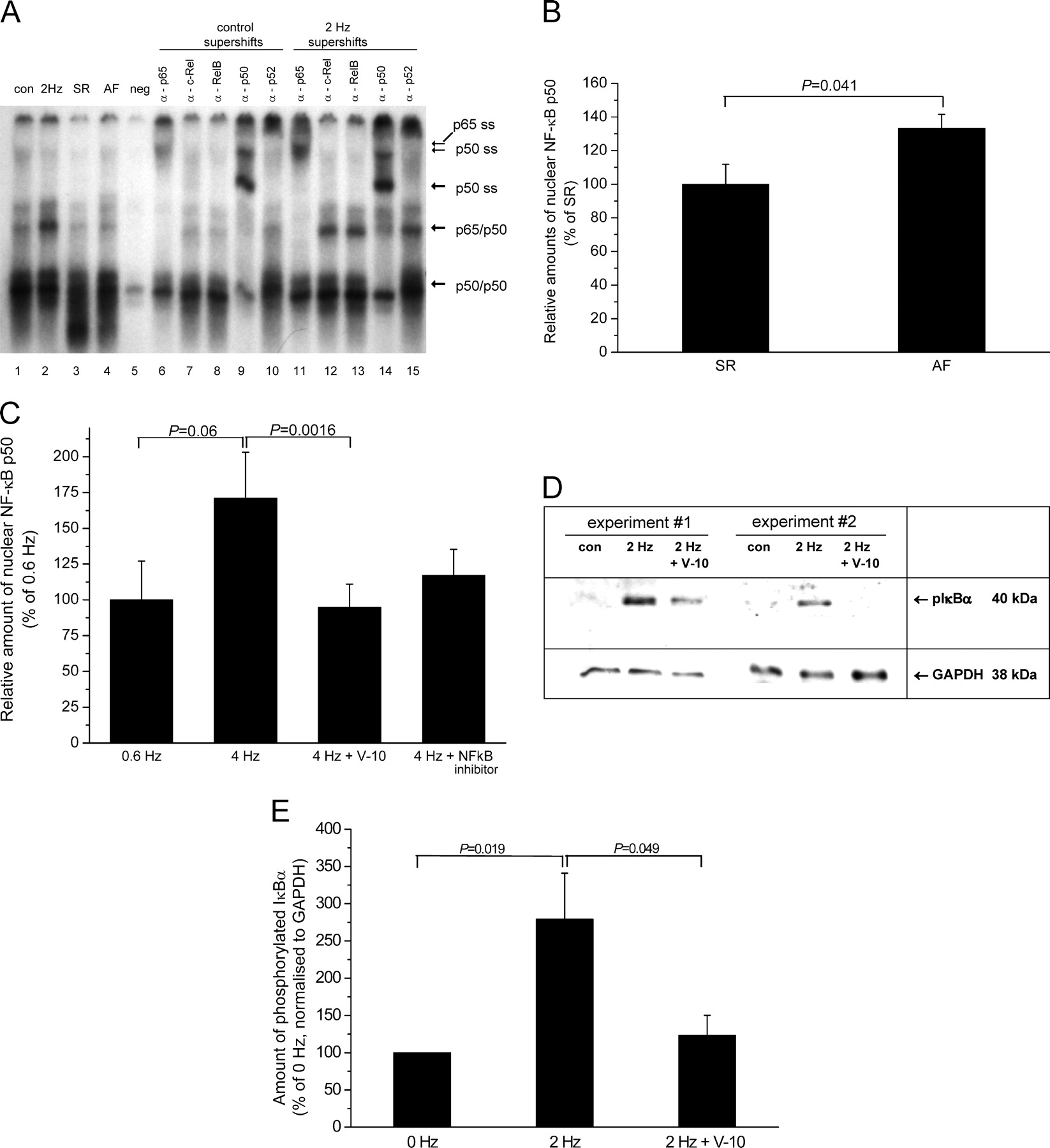
Activation of NF-κB. (A) Increased nuclear amounts of NF-κB were observed in atrial tissue slices after rapid pacing (2 Hz vs. control; lanes 1, 2) and in atrial tissue from patients with AF (lane 4 vs. SR lane 3). Supershift (SS) was achieved with anti-p50 and anti-p65 antibodies (lanes 6, 9, 11, and 14), indicative of the classical NF-κB pathway being involved (neg. = excess unlabelled oligonucleotide). (B and C) Increased nuclear amounts of NF-κB p50 were detected by ELISA in ex vivo atrial tissue from patients with AF in comparison with SR (B) and in response to rapid pacing in vitro compared with pacing at 0.6 Hz (C). Verapamil (10 μM) effectively prevented the nuclear accumulation of p50 in response to rapid pacing, whereas the NF-κB inhibitor IV (Merck, Darmstadt, Germany) showed a similar, but less pronounced effect (C) (n = 4). 100% is equivalent to 0.315 ng p50/μg nuclear protein in B and to 0.346 ng p50/μg nuclear protein in C. (D) Representative immunoblot (40 μg of protein per lane) showing phosphorylated IκBα. (E) Quantitative analysis of the blots as shown in D revealed a nearly 3-fold increase of phosphorylated IκBα after pacing. Verapamil (10 μM) reduced phospho-IκBα levels to almost baseline vs. 2 Hz (n = 5).
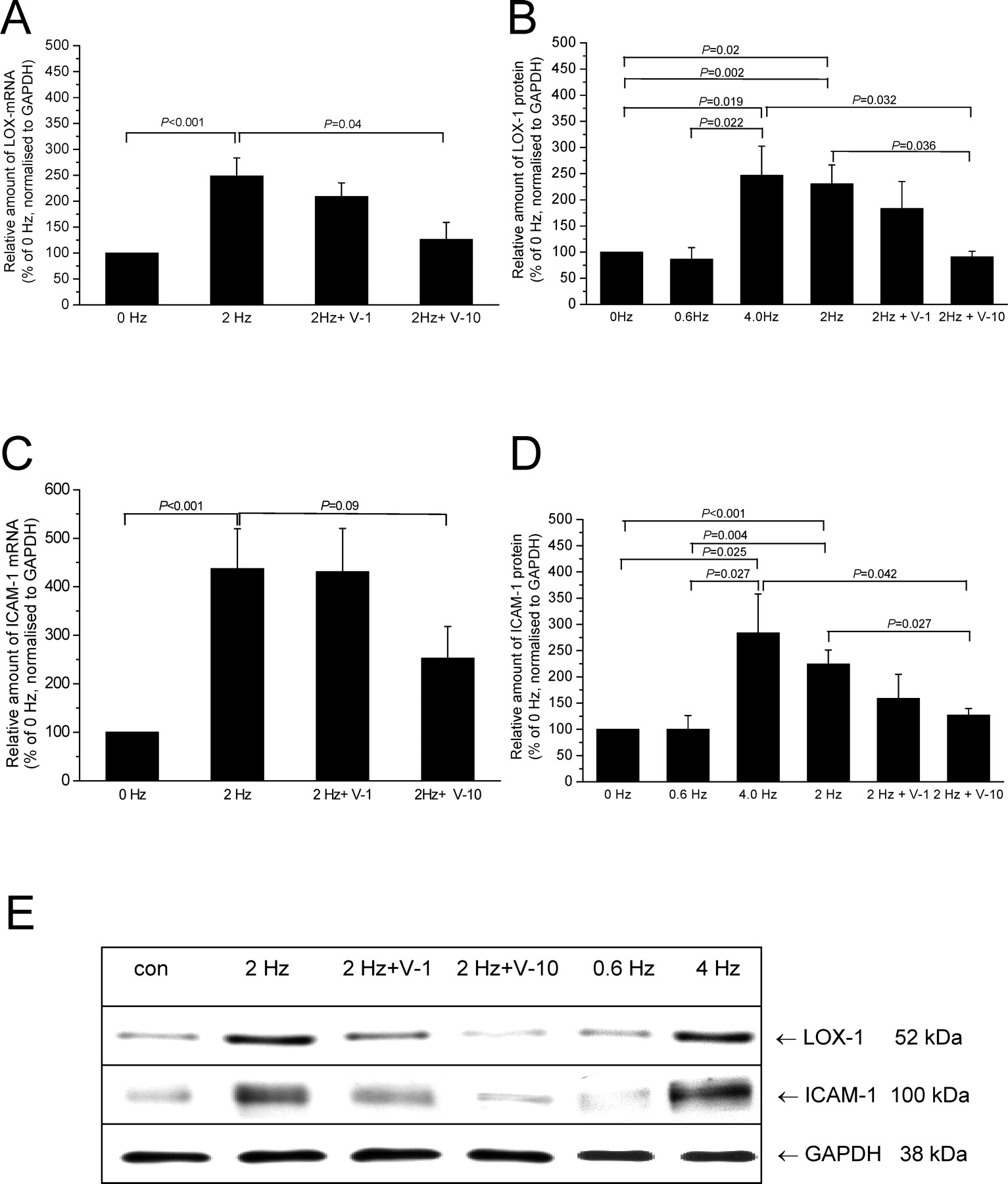
Induction of the NF-κB target genes, LOX-1 and ICAM-1, in response to rapid pacing. Atrial tissue slices were paced at the frequencies indicated in the absence/presence of 1–10 μM verapamil for 24 hrs. Rapid pacing increased amounts of LOX-1 (A) and ICAM-1 (C) mRNA significantly. Verapamil dose-dependently decreased these elevated LOX-1 and ICAM-1 mRNA amounts, but statistical significance was observed at 10 μM verapamil only. LOX-1 (B) and ICAM-1 (D) protein amounts were significantly increased in response to rapid pacing (2 or 4 Hz) when compared with atrial tissue slices paced at 0 (non-paced control) or 0.6 Hz. Again, verapamil prevented in a dose-dependent manner this increase in LOX-1 and ICAM-1 expression (B and D). (E) Representative immunoblots showing LOX-1 and ICAM-1 expression in human atrial tissue. GAPDH was used as loading control.
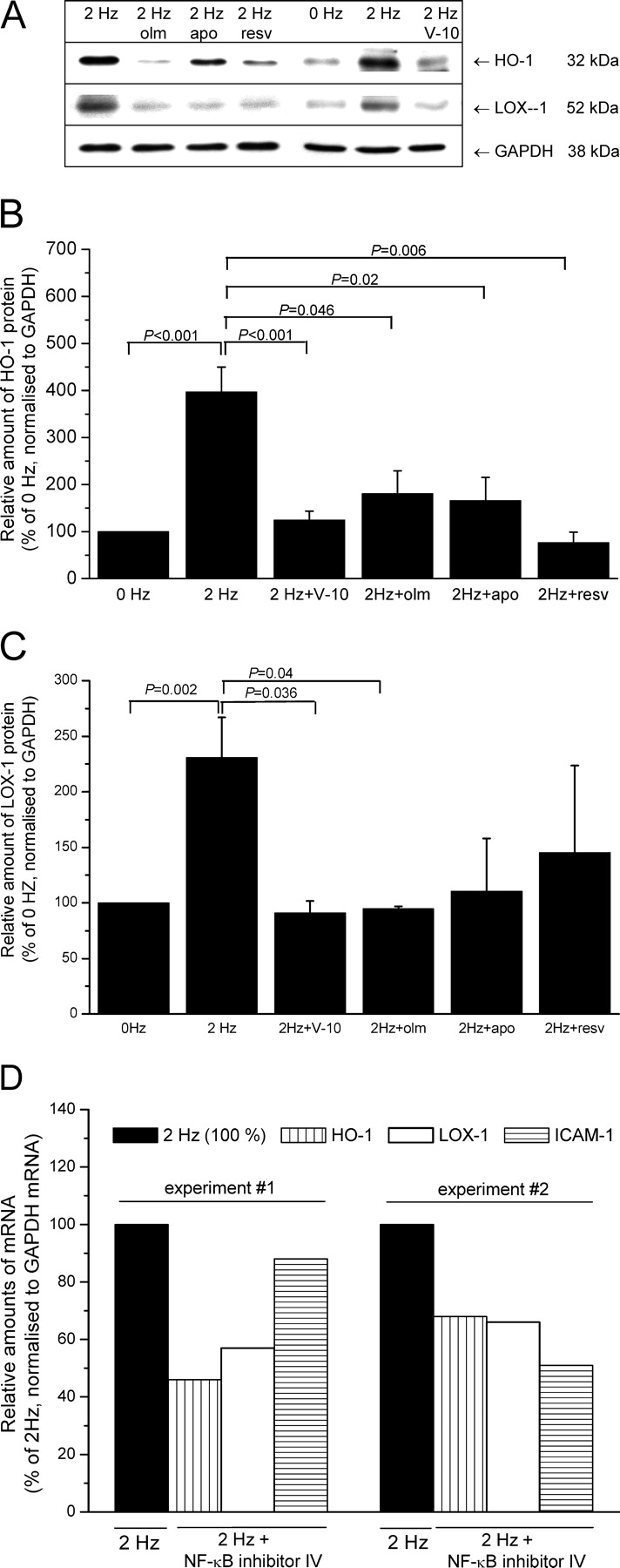
Effects of inhibitory compounds on the pacing-induced expression of NF-κB target genes, LOX-1 and HO-1. (A) Representative immunoblot demonstrating the capability of verapamil (V-10), olmesartan (olm), apocynin (apo), and resveratrol (resv) to prevent the pacing-dependent increase in LOX-1 and HO-1 protein. (B, C) Quantitative analyses of blots as shown in A revealed that all compounds significantly diminished HO-1 (B), whereas only verapamil and olmesartan significantly reduced LOX-1 (C) (n = 3). (D) As expected, two independent experiments demonstrated that the administration of NF-κB inhibitor IV (Merck) negatively affects the pacing-dependent induction of transcription of NF-κB target genes HO-1, LOX-1, and ICAM-1.
Footnotes
1
These authors contributed equally to this paper.
This work was supported by the “Bundesministerium für Bildung und Forschung, Germany” (Kompetenznetz Vorhofflimmern, 01GI0204).
Acknowledgements
We thank Katja Mook and Doris Trzeczak for excellent technical assistance.