
Abstract
Fibrogenesis is a mechanism of wound healing and repair. However, prolonged injury causes deregulation of normal processes and results in extensive deposition of extracellular matrix (ECM) proteins and fibrosis. The current review will discuss similarities and differences of fibrogenesis in different organs and systems and focus on the origin of collagen producing cells. Although the relative contribution will vary in different tissues and different injuries, there are three general sources of fibrogenic cells: endogenous fibroblasts or fibroblast-like cells, epithelial to mesenchymal transition, and recruitment of fibrocytes from the bone marrow.
Fibrosis is a common pathophysiological response of many tissues to chronic injury. On the one hand, wound healing, tissue remodeling and repair are protective mechanisms activated in response to stress and injury in order to maintain functional integrity of organs and systems. On the other hand, deregulation of normal healing and continued exposure to chronic injury results in tissue fibrosis, massive deposition of extracellular matrix, scar formation, and organ failure. This review will focus on the pathogenesis and common features of fibrosis in different tissues. In particular, the source of fibrogenic cells contributing to tissue scarring will be discussed.
Fibrosis is caused by a series of events, triggered by chronic injury. These events include: 1) immediate damage to the epithelial/endothelial barrier; 2) release of TGF-β1, the major fibrogenic cytokine; 2) recruitment of inflammatory cells; 4) induction of reactive oxygen species (ROS); 5) activation of collagen producing cells; 6) matrix activation of myofibroblasts; and, finally, 7) in the absence of continuous injury, reversal of fibrosis (Table 1) can occur. All listed factors play a significant role in fibrogenesis; however, release of TGF-β1 is the key event in pathogenesis of fibrosis. All factors are complimentary to each other and synergistically contribute to the development of fibrosis. The specific role of each event in each organ needs to be determined.
Damage to Epithelial/Endothelial Barrier
Vascular damage plays an important role in fibrogenesis. It is one of the leading events in pathogenesis of systemic sclerosis, which contributes to the vascular thickening, production of pro-inflammatory cytokines and TGF-β1, tissue hypoxia, platelet aggregation, decreased nitric oxide and lead to the development of fibrosis (1, 2). It occurs at the microvascular level (single EC cell) and/or macrovascular level (tissue vasculature) (3). There are two effects of endothelial damage; acute (coagulation and vasodilation) and delayed (neovascularization of fibrotic areas) (3). In addition, EC can also contribute to fibrosis by endothelial-to-mesenchymal transition (4, 5), see chapter EMT).
Vascular Damage and Vasculogenesis.
Vascular damage is critical for the onset of fibrosis. Disruption of the integrity of the endothelium results in activation of an anti-fibrinolytic cascade to support coagulation and hemostasis (6, 7). Endothelial cells (EC) start secreting factors, which induce platelet aggregation and degranulation, blood clot formation, and accumulation of provisional ECM. Following platelets degranulation, released vasodilating cytokines increase permeability of the blood vessels and loss of barrier function with lymphocyte migration through the endothelial barrier. Vascular damage fibrosis is further facilitated by damage of the blood vessel basement membrane and rapid recruitment of inflammatory cells to the site of injury (8). Although vasculogenesis is closely linked to inflammation, it is a distinct mechanism, and EC themselves secrete cytokines to actively recruit inflammatory cells, e.g., neutrophils and macrophages. Macrophages do not only phagocytose products of degradation and cellular debris, but produce fibrogenic signals and secrete factors chemotactic for EC. In turn, EC proliferate and migrate through the basal membrane toward the center of the wound to facilitate healing. In chronic injury, EC are involved in vascular remodeling, and together with circulating endothelial progenitor cells grow new blood vessels into the damaged tissue (9, 10). Vasculogenesis is often observed in cancer associated fibrosis: Damaged tissues compensate their high demand for nutritional supplies by extending the capillary net from the pre-existing vasculature into fibrotic areas (11). Vasculogenesis is strictly regulated on several levels, including EC proliferation, EC migration, and tube formation. Numerous mediators of angiogenesis, including such fibrogenic factors as angiopoietins, transforming growth factor (TGF-β1), platelet-derived growth factor (PDGF), tumor necrosis factor-alpha (TNF-α), interleukins, and members of the fibroblast growth factor family (FGF), have been identified (12). However, vascular endothelial growth factor (VEGF) remains the strongest angiogenic inducer (3). It controls EC survival, proliferation, and postnatal angiogenesis. VEGF binds to the receptor VEGFR2 and mediates its biological responses through reactive oxygen species (ROS) (13). Another strong angiogenic factor, Angiopoietin 1 (Ang 1), regulates maturation and stability of blood vessels. It signals through endothelial receptor tyrosine kinase Tie2 and synergistically enhances VEGF’s effects (14). Activation and vascular maturation along with important cytoskeletal rearrangements in EC are regulated by sphingosine 1-phosphate (S1P), a biologically active lipid growth factor, which binds cell surface receptor-mediated G protein and signals through the Rho family of small GTPases (15). Finally, ELR+ motif (Glu-Leu-Arg domain) containing CXC chemokines provide a unique regulatory mechanism of angiogenesis to endorse angioproliferative signals in EC. Angiogenic chemokines include CXCL1, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, and CXCL8, and utilize mostly CXCR2 and CXCR3 receptors to modulate their effects (11).
Apoptosis of Epithelial Cells.
Epithelial cells and EC are located in close proximity, and injury of the endothelium and the basal membrane often affects the epithelium. Similar to EC, damaged epithelial cells also contribute to inflammation and fibrogenesis. They become activated, secrete cytokines, growth factors, and chemo-attractants for mononuclear cells and interstitial fibroblasts, and apoptose or transdifferentiate into fibroblasts. In addition, epithelial cells can facilitate injury of endothelium and promote angiogenesis [reviewed in (16)]. Apoptosis of damaged epithelium is caused by several common mechanisms: 1) death receptor-mediated pathway [Fas-FasL interaction, (17, 18)]; 2) loss of contact to the basement membrane; 3) intrinsic organelle-dependent pathway, which involves lysosomal permeabilization, or release of cathepsin B into cytoplasm and mitochondria damage (16, 19, 20).
Apoptosis is a part of normal tissue homeostasis and repair. Thus, apoptosis, phagocytosis of the dead cells, and subsequent clearance of macrophages are the normal pathway for the resolution of dead cells. Removal of myofibroblasts by programmed cell death is required for the normal resolution of tissue repair responses (21). Dysregulation of this process, such as resistance of myofibroblasts or inflammatory cells to apoptosis, may result in a chronic inflammation or fibrosis. Moreover, exaggerated apoptosis of epithelial cells also causes deranged tissue homeostasis and contributes to fibrosis. Dead epithelial cells form apoptotic bodies, which are then phagocytosed by stimulated macrophages (19). Apoptosing epithelial cells secrete cytokines, which recruit macrophages and induce their activation. Activated macrophages release TGF-β1 and in turn promote proliferation and activation of collagen producing cells (22). Activated myofibroblasts can, in turn, produce a high amount of TGF-β1 and of extracellular ROS, specifically hydrogen peroxide, and further facilitate apoptosis of damaged epithelial cells (19, 23). Similar responses to injury have been described for pulmonary epithelium (alveolar pneumocytes), kidney epithelium, and hepatic epithelium (hepatocytes) (18, 19, 21). Thus, induction of alveolar epithelium apoptosis by anti-Fas (activating) antibody is associated with the development of pulmonary fibrosis in mice (24). Liver hepatocytes are more susceptible than other cells to toxic reagents, including alcohol, bile acids, and viral infection. They release reactive oxygen species (ROS) and secrete fibrogenic factors (CXC chemokines, KC, and MIP-2), which activate collagen-producing cells and inflammatory cells, and are the first cells to undergo apoptosis in response to irreversible injury (19).
Damage to lung epithelial cells is associated with interstitial edema, leukocyte invasion, and decreased gas exchange, while chronic dysfunction of tubular epithelium often leads to renal dysfunction, tubular atrophy, and development of progressive renal disease (16, 20). Damage of endothelial/epithelial barrier and apoptosis of epithelial cells results in inflammation and activation of Toll-like receptor (TLR) signaling, which is associated with release of fibrogenic cytokines in the injured tissues (25).
Release of TGF-β1
The cytokine transforming growth factor-β1 (TGF-β1) promotes wound healing and repair. Under pathological conditions, TGF-β1 orchestrates a cross talk between parenchymal, inflammatory, and collagen-expressing cells and plays a key role in stimulating fibrosis. Overexpression of TGF-β1 in transgenic mice results in fibrosis of multiple organs, suggesting that TGF-β1 is a major pro-fibrogenic cytokine (19). TGF-β1 is synthesized by different cell types in different organs. In damaged liver, apoptotic hepatocytes release some TGF-β1; however, Kupffer cells are the major source of TGF-β1. TGF-β1 released by Kupffer cells is critical for activation of hepatic stellate cells (HSCs), the primary source of collagen type I in fibrotic liver (19). Once activated, HSCs and sinusoidal endothelial cells also contribute to TGF-β1 production (19). During pulmonary fibrosis, monocytes, eosinophils, myofibroblasts, and fibroblasts secrete TGF-β1 (11).
To mediate its function, TGF-β1 undergoes several important post-translational modifications. TGF-β1 is synthesized as a non-active proform, cleaved intracellularly by endopeptidase furin to generate mature form, but remains biologically inactive due to association with two proteins: latency-associated peptide, LAP, and latent TGF-β binding protein, LTBP. This large TGF-β1-associated complex is then secreted into extracellular matrix, where it is cross-linked by tissue transglutaminase and stored as a reservoir without any effect on tissues (26). Activation of mature TGF-β1 requires release from latency maintaining protein complex LAP/LTBP.
TGF-β1 is activated by binding to thrombospondin 1 (TSP-1) or ανβ6 integrin, acidification, or proteolysis (26–30). TSP-1, a large glycoprotein, is implicated in angiogenesis, cell adhesion, and remodeling of ECM (30, 31). Integrin ανβ6, a heterodimeric matrix receptor in epithelial cells, some dendritic cells, and macrophages, together with other ανβ members, mediate cell adhesion, migration, and connect cytoskeletal proteins to ECM (32). It is not expressed constitutively but is strongly upregulated during injury and inflammation (26). It binds to the tri-peptide recognition sequence arginine-glycine-aspartic acid (RGD) in its ligands: fibronectin, tenascin-C, vitronectin, and the LAP of TGFβ1. Direct binding of ανβ6 to RGD sequence in LAP results in TGF-β1 activation (29). ανβ6-dependent presentation of the latent complex to MMP-14 is critical for LAP degradation and release of TGF-β1 (26). It has been also suggested that association of ανβ6 integrin with LTBP-1 causes mechanical traction important for activation of latent TGF-β1 (32). Alternatively, TGF-β1 can be activated by several proteases such as plasmin or matrix metal-loproteinases MMP-2 and 9, which directly induce degradation of the LAP/LTBP complex (33). Neutrophil elastase, a serine protease released by neutrophil degranulation, has also been implicated in activation of latent TGF-β1 (34). Although the mechanisms of latent TGF-β1 activation into the biologically active form may vary in different organs and tissues, the role of TGF-β1 in pathogenesis of fibrosis is indispensable. Release of TGF-β1 is strongly required for fibrogenesis but alone is not sufficient to develop fibrosis. Other factors, discussed in this review, contribute to ECM deposition and fibrogenesis.
Subsequently, activated TGF-β1 modulates its biological effects through binding to specific TGF-β1 receptors (TβR). There are three TGF-β1 receptors: TGF-β receptor type I (TβRI), TGF-β receptor type II (TβRII), and TGF-β receptor type II (TβRIII) (28). Both TβRI and TβRII are the transmembrane receptors with serine–threonine kinase activity, and they are present as homodimers in the plasma membrane (35). TGF-β1 binds primarily TβRII. Ligand binding induces the assembly of type I and type II receptors into complexes, within which TβRII phosphorylates and activates TβRI. This phosphorylation event results in activation of TβRI kinase and subsequent downstream signaling (36, 37). Binding of TGF-β1 to TβRI, an effector protein, determines specificity of the signaling. The third receptor for TGF-β1 (TβRIII) consists of the accessory proteins (betaglycan and endoglin), and facilitates delivery of the ligand to the TβRII receptor (28). TGF-β1 signals are mediated by Smad (composite name from Sma [Caenorhabditis elegans] and Mad [Drosophila melanogaster]) family of molecules, comprised by three distinct classes: 1) receptor-regulated Smads (R-Smads), which include Smad1, 2, 3, 5, and 8; 2) common-mediator (co-Smad) Smad4; and 3) antagonistic or inhibitory Smads, Smad6 and 7. The growing evidence suggests that Smad3 is critical for fibrogenesis of lung, liver, and kidney (38–40). The role of Smad2 in fibrosis is less characterized since Smad2 deletion is lethal, but seems distinct from Smad3 due to regulation of different target genes (41). Activated Smad2/3 complex forms hetero-oligomers with another member of the Smad family, Smad4. In association with Smad4, Smad2/3 complex is translocated to the nucleus, where it initiates transcription of TGF-β1 target genes. This pathway is regulated by several autoinhibitory feedback loops. Smad7 is an inhibitor of TGF-β1 that is expressed in response to prolonged TGF-β1 signaling; it binds to TβRI and abrogates TGF-β1 effects. In addition, Ski, SnoN, and Bambi are negative regulators of TGF-β1 signaling (28, 42).
Connective tissue growth factor (CTGF) is another important fibrogenic factor. CTGF is a downstream mediator of TGF-β1 responses synthesized in the Smad3-dependent manner (43). Two mechanisms underlie the synergistic effect of CTGF on TGF-β1. First, CTGF acts as a chaperone that augments TGF-β1 signaling; it directly binds TGF-β1 and increases its affinity to TGF-β1 receptors, causing a sustain enhanced response (33). Second, CTGF blocks a TGF-β1 auto-inhibitory feedback loop, in that CTGF suppresses de novo synthesis of Smad7 via activation of transcription factor TIEG-1 (44). These mechanisms of CTGF signaling have been documented in fibrogenesis in skin, lungs, liver, and kidneys (43). In addition, CTGF regulates cellular apoptosis and fibroblast proliferation, angiogenesis, cellular adhesion, chemotaxis, and deposition of ECM (43). Although the CTGF-specific receptor is unknown, integrins and heparin sulfate proteoglycans mediate many of CTGF functions (33).
Platelet-derived growth factor (PDGF) is a potent fibrogenic growth factor known to synergize with TGF-β1 (28). PDGF is secreted by platelets, tissue macrophages, activated fibroblasts, myofibroblasts, and HSCs and signals through PDGFR-α and -β, a receptor with intrinsic tyrosine kinase activity. There are four isoforms of PDGF (PDGF-A, B, C, and D) that are implicated in liver, lung, and kidney fibrosis (45). PDGF induces migration and proliferation of mesenchymal cells to the site of injury (45). Binding of PDGF to its receptors induces receptor auto-phosphorylation and activation of MEK/ERK and PI3K/AKT signaling pathway (46). Hepatocyte growth factor (HGF) acts as an endogenous anti-fibrotic factor and has potential as an anti-fibrotic therapy (47). Despite its name, HGF is widely expressed in many tissues in response to elevated levels of TGF-β1 (48). HGF possess anti-fibrogenic, anti-inflammatory, and pro-regenerative properties and strongly inhibits myofibroblastic differentiation (49). HGF antagonizes TGF-β1 signaling by interfering with nuclear translocation of TGF-β1–activated Smad proteins or induction of TGF-β1 co-repressor SnoN (50). However, TGF-β1 and HGF mutually inhibit each other, and high levels of TGF-β1 may dominate over HGF effects. Meanwhile, the anti-inflammatory function of HGF is mediated through another pathway. HGF-mediated inhibition of NF-κB signaling induces down-regulation of pro-inflammatory cytokines (TNF-α, IFN-γ, MCP-1, IL-12) and prevents recruitment of inflammatory cells to the injured organs (49).
Inflammation
Inflammation usually precedes fibrosis. Recruitment of inflammatory cells to the site of the acute injury is a part of wound healing, and TGF-β1 is a potent chemoattractant for cells of macrophage-monocytic lineage. In addition to TGF-β1, MCP-1, MIP-2, and MIP-1 are also involved in recruitment of inflammatory cells (19). Initial inflammation is caused by cytokine-mediated endocytosis/phagocytosis. Neutrophils are the first cells recruited, they uptake cell debris and phagocytose apoptotic bodies. Activated neutrophils degranulate, release inflammatory and pro-fibrogenic cytokines, and apoptose. Following neutrophils, macrophages infiltrate damaged tissues, phagocytose, and secrete fibrogenic cytokines. Macrophages are a major source of TGF-β1 in fibrosing organs. T and B lymphocytes are also recruited to the site of injury and further facilitate secretion of fibrogenic cytokines.
Pulmonary inflammation promotes fibrosis, induces fibroblast and fibrocyte proliferation, and ECM deposition (51). In the skin, migration of inflammatory cells to the wound is triggered by TGF-β1, released by macrophages. In addition to cells recruited from blood, there is a small population of endogenous skin mast cells secreting TGF-β1 (52). In renal fibrosis, the activation of the reninangiotensin system (RAS) and its main effector angiotensin II (Ang II) stimulates inflammation, including the expression of cytokines, chemokines, growth factors, and reactive oxygen species (53). Ang II induces vascular inflammation, endothelial dysfunction, upregulation of adhesion molecules, and recruitment of infiltrating cells into the kidney (54). Myelomonocytic cells produce most of the TGF-β1 in injured kidney. In the liver, macrophages derive from two different sources (bone marrow macrophages and liver resident Kupffer cells) and play a critical role in promotion and resolution of hepatic fibrosis. Dual effect of liver macrophages on liver fibrosis was demonstrated in mice devoid of CD11b+ myelomonocytic cells, including macrophages and some Kupffer cells. These mice developed less fibrosis but exhibited delayed resolution of fibrosis (55). Our studies also suggested that Kupffer cells are required for fibrosis (unpublished observations).
The role of bacterial flora and toll-like receptors (TLRs) in the pathogenesis of fibrosis has been recently demonstrated. Toll-like receptors (TLRs) are innate immune signal receptors which recognize pathogen-associated molecular patterns (PAMP) such as lipopolysaccharide (LPS), gram-negative bacterial cell wall component, peptideglycan, gram-positive bacterial cell wall component, and bacterial-derived unmethylated CpG-DNA. In addition, endogenous ligands (alarmins) can bind TLR4 in the presence of CD14 and LPS binding protein (LBP) and transduce similar signals (56). Thus, endogenous ligand HMGB-1, hyaluronan, and products of dying cells have been shown to trigger TLR signaling. Upon activation of TLRs, cells produce pro-inflammatory cytokines, such as TNF-α, IL-6, IL-1, MCP-1, and RANTES. Activation of TLR2 and TLR4 signaling was reported in lung and renal injury (57, 58). Moreover, microbial products have a significant impact on fibrogenic progression, and LPS synergistically facilitates other fibrogenic factors such as TGF-β1, oxidative stress, and mechanical injury. In the lungs, continuous mechanical stretch of alveolar epithelium by forced ventilation results in pulmonary injury and fibrosis. This process is significantly exacerbated in patients and experimental models in mice by concomitant bacterial infections (59). It is proposed that bacterial products interact with lung epithelial cells and induce intracellular signaling to secrete fibrogenic cytokines. TLRs are also critical in liver fibrosis (60). Patients with hepatic cirrhosis have elevated portal vein levels of LPS. High portal vein pressures can damage intestinal mucosa and compromise its barrier function and trigger bacterial translocation. Therefore, LPS derived from the intestinal microflora is a strong candidate for the TLR4 ligand in hepatic fibrosis (61).
Oxidative Stress
Oxidative stress, caused by increase in reactive oxygen species (ROS), such as superoxide, hydrogen peroxide, and hydroxyl radicals, is closely associated with fibrosis (19). Decreasing levels of endogenous antioxidants further contribute to tissue damage. In the liver
Moreover, in activated and proliferated collagen producing cells, ROS acts as an intracellular second messenger. Thus, angiotensin II (Ang II) stimulates DNA synthesis, cell migration, procollagen alpha1(I) mRNA expression, and secretion of TGF-β1 and inflammatory cytokines in hepatic stellate cells. These Ang II–induced profibrotic actions are largely mediated by ROS generated by a nonphagocytic form of NADPH oxidase in hepatic stellate cells. Consistent with this, NADPH oxidase p47phox-null mice are resistant to experimental liver fibrosis (67). PDGF also stimulate intracellular ROS production via NADPH oxidase in hepatic stellate cells (68). ROS is involved in fibrogenesis in kidneys and is a key factor in glomerulosclerosis and tubulointerstitial fibrosis. Renal ROS are produced by fibroblasts, endothelial cells (EC), vascular smooth muscle cells (VSMC), mesangial cells (MCs), tubular cells, and podocytes (69). Anti-oxidants are effective in inhibiting many models of fibrosis but have not been effective in clinical trials in patients.
Activation of Collagen-Producing Cells
Resident Myofibroblasts.
A growing body of evidence from studies conducted in different organs suggests that resident myofibroblasts are the primary source of ECM in the course of fibrosis (Table 2). Resident myofibroblasts arise from a population of tissue specific fibroblasts that proliferate and undergo activation in response to injury (Fig. 1). Classical myofibroblasts differentiate from a mesenchymal lineage and, therefore, lack expression of lymphoid markers such as CD45 or CD34. In the presence of fibrogenic stimuli in culture, such as TGF-β1 or certain types of ECM (fibronectin, collagen type I), they rapidly acquire a myofibroblastic phenotype, including expression of α-SMA, secretion of collagen type I and III, and contractility [Fig. 1; (6)]. Resident fibroblasts/myofibroblasts are believed to be the major source of collagen producing cells in lungs, skin, kidneys, and liver.
Thus, in fibrotic lungs the majority of myofibroblasts arise from pre-existing fibroblasts such as peribroncholiolar and perivascular adventitial fibroblasts. In response to fibrogenic stimuli, they upregulate α-SMA, vimentin, collagen, often secrete TGF-β1 and MCP-1, and migrate (10, 70, 71). Similarly, dermal fibroblasts give rise to a population of myofibroblasts and induce scarring of injured skin (72, 73). In diseased kidneys, activation of cortical fibroblasts into myofibroblasts correlates with development of tubulointerstitial fibrosis (74). Normally cortical fibroblasts comprise a population of quiescent cells with a low turnover rate, but they proliferate, produce excess of matrix proteins, and become principal mediators of fibrosis in response to injury (75, 76). In fibrotic livers, the primary collagen producing cells originate in the liver but possess unique features. The majority of liver myofibroblasts arises not from fibroblasts, as it has been demonstrated for other organs, but from hepatic stellate cells (HSCs). In normal physiology, HSCs have a quiescent phenotype and regulate vitamin A homeostasis (77). Quiescent HSCs express neural markers, such as GFAP, synamin, synaptophysin, and nerve growth factor receptor p75 (78). In response to injury, quiescent HSCs undergo morphological and functional changes, lose vitamin A expression, and activate into myofibroblasts, expressing the typical myofibroblast markers (collagen type I, α-SMA, desmin, vimentin, and contractility), and are capable of phagocytosis and antigen presentation (67, 79). Portal fibroblasts are another population of endogenous liver cells implicated in hepatic fibrogenesis (80), reviewed in (19, 81, 82). Derived from small portal vessels, they express markers distinct from HSCs. Induced by cholestatic liver injury, portal fibroblasts proliferate and deposit collagen around biliary tracts (83). However, activation of portal fibroblasts occurs mainly during portal injury, they proliferate more slowly than activated HSCs, and probably have a smaller contribution to liver fibrosis.
Several other tissue resident cell types also contribute to fibrosis (73) Pericytes and vascular smooth muscle cells (SMC) are an additional source of local myofibroblasts in scleroderma, liver, and glomerular fibrosis (84, 85). Consistent with this, mesangial cells (kidney pericytes) undergo activation from quiescence to fibroblastic, proliferate, and produce IL-1, MCP-1, RANTES, and TGF-β1 and contribute to renal fibrosis (86).
Epithelial-to-Mesenchymal Transition (EMT).
Recent studies suggest that in response to injury, epithelium can contribute to fibrosis by generating fibroblasts via epithelial-to-mesenchymal transition (EMT). EMT has also been reported in localized and metastatic cancer. EMT is “a process when fully differentiated epithelial cells undergo phenotypic transition to fully differentiated mesenchymal cells (fibroblasts or myofibroblasts)” [reviewed in (87)]. There are fundamental differences between epithelial and mesenchymal cells. Epithelium is formed by a monolayer of polarized cells, which are strictly organized, regularly spaced, connected by tight cell-cell junctions and adhesions, and devoid of individual cell movement (88). In contrast, mesenchymal cells exhibit high motility, have elongated shape, and lack rigid intracellular structure or tight intercellular connections (88). Cellular conversion from epithelial to mesenchymal phenotype requires a dramatic alteration of transcriptional machinery and reprogramming of cellular morphology, architecture, adhesion, and migratory capacity (88). These changes are attributed to high plasticity of epithelial cells. Therefore, epithelium is considered a multipotent progenitor tissue, which can undertake alternative developmental pathways in response to injury: re-epithelialization to restore normal tissue, apoptosis, or fibrosis via EMT. EMT occurs physiologically during embryonic development (89). In adult tissues, EMT represents a rare event during wound healing. Evidence of EMT in adult tissues has been reported during fibrosis in the lungs, kidneys, and liver.
Several steps are critical for EMT: 1) disruption of local basement membrane; 2) loss of epithelial cell adhesion; 3) reprogramming of signaling machinery and de novo synthesis of α-SMA; and 4) rearrangement of cytoskeletal proteins and transmigration of epithelial cell through the basement membrane into interstitial space (75, 90–92). The onset of EMT requires contact injury and release of TGF-β1 (90). Damage of basal membrane (loss of integrity or proteolytic degradation by MMPs) and release of growth factors trigger and further facilitate EMT. Although secretion of TGF-β1 plays a key role in EMT progression (92), other factors such as epithelial growth factor (EGF), basic fibroblasts growth factor (bFGF), and interleukin-1 (IL-1) also contribute. Epithelial cells normally line up along basement membranes and are tightly connected to each other through intercellular adhesion molecules like E-cadherin. In response to chronic injury, epithelial cells undergo activation, detach from damaged basement membranes, and start secreting the EMT-promoting cytokines (92). They also lose expression of epithelial markers (e.g., E-cadherin and zonula occludens-1, ZO-1), and change their polygonal shape and apical-basal polarity to become elongated. Such epithelial cells start expressing fibroblast-specific protein (FSP-1), extracellular matrix proteins (e.g., fibronectin, collagen type I and III), and α-smooth muscle actin (α-SMA) (Fig. 1). They attain high contractility and migrate through the basement membrane into the interstitium, where they proliferate and give rise to collagen secreting myofibroblasts [reviewed in (90)].
EMT is best characterized in kidneys. It is estimated that one third of renal interstitial fibroblasts, the main mediator of renal interstitial fibrosis, are derived from tubular epithelial cells via EMT (93). Renal epithelial cells have unique characteristics. Unlike epithelium in other parenchymal organs, which develops from primary epithelial sheets, renal epithelium have two distinct embryonic origins: the mesenchymal blastema and epithelial Wolffian duct (94, 95). Most of the renal epithelium undergoing EMT is of mesenchymal origin (92, 94). The greater plasticity of renal epithelium may result in the significant contribution of EMT to renal fibrosis.
EMT has also been implicated in pathogenesis of pulmonary fibrosis. Alveolar epithelial cells (AEC) (10) are the source of EMT in injured lungs. Alveolar epithelium is composed of two types of epithelial cells; elongated alveolar type I (ATI) cells, responsible for the gas exchange, and cuboidal alveolar type II (ATII) cells, which synthesize and secrete surfactant (96). ATII cells may be pulmonary multipotent progenitors capable of self renewal and proliferation, differentiation into ATI cells and into fibroblasts to repair injured AEC. TGFβ is induced in response to injury and activates ATII that may differentiate into ATI cells to induce re-epithelialization, apoptosis, or undergo EMT (97). In lung fibrosis, both ATII and ATI change morphology and gene expression patterns, down-regulate markers of pulmonary epithelium markers (e.g., cytokeratins, surfactant protein C, thyroid transcription factor [TTF]-1), produce TGF-β1, and upregulate fibroblast-specific markers (97).
EMT may also occur in the liver. Fetal liver exhibit characteristics of EMT in that some fibroblast-like stromal cells co-express both epithelial (α-fetoprotein [AFP], albumin [Alb], cytokeratins CK18 and CK7) and mesenchymal markers (α-SMA, osteopontin, and collagen I) (98–100). In adult liver, EMT does not occurs without stress or injury (98). Transformation of biliary epithelium into fibroblasts is best documented in metastatic hepatocellular carcinoma, but not hepatic fibrosis. Evidence of EMT in liver fibrosis was reported in a patient with primary biliary cirrhosis (PBC), a condition characterized by loss of biliary epithelial cells and progressive fibrosis (101). Analysis of liver biopsies from this patient revealed that a number of biliary epithelial cells expressed markers of EMT (e.g., an early fibroblast marker FSP1, vimentin, nuclear Smad2/3 and α-SMA), suggesting that biliary epithelial cells may undergo EMT and potentially contribute to the fibroblast population (101). In mice, EMT was observed in response to bile duct ligation (BDL)–induced injury (102). BDL causes chronic obstruction and concomitant proliferation of the bile ducts, outgrowth of periductal myofibroblasts and fibrosis. Expression of EMT markers (collagen type I, α-SMA, and cytokeratin 19) by periductal myofibroblasts supported a notion that biliary epithelial cells may undergo EMT (102). Another cell type potentially capable of EMT in the liver is the oval cell. Oval cells comprise a small population of bipotential progenitor cells, which exhibit high plasticity and give rise to hepatocytes and cholangiocytes (103).
TGF-β1, the main mediator of EMT, signals through classical Smad3-dependent and/or Smad3-independent pathways, which collaboratively reinforce EMT. Activation of Smad3 results in downregulation of E-cadherin, loss of adhesion, and increased cell motility via recruitment of integrin-linked kinase (ILK)-β-catenin signaling [reviewed in (90, 93)]. Activation of non–Smad-mediated signaling by TGF-β1 requires Rho kinase, which directly induces EMT genes [cytoskeletal rearrangement, E-cadherin downregulation, detachment of basement membrane (104)]. Thus, TGF-β1 binding to its epithelial receptor activates Ras, which in turn activates Rho and Raf/MAP kinases to cause reorganization of cytoskeletal actins (90, 104–106). Triggered by contact disassembly, activated Rho and Rho kinase (ROK) mediate nuclear translocation of serum response factor (SRF) and its coactivator, myocardin-related transcription factor (MRTF), which directly activate the α-SMA promoter (107).
Moreover, two other signals also facilitate EMT: 1) TGF-β1–induced apoptosis of epithelial cells, which do not undergo EMT and 2) expression of fibroblast-specific protein 1 (FSP1) by epithelial cells acquiring a myofibroblast phenotype (92, 93, 108). FSP1, an intracellular calcium-binding protein, is induced in newly formed fibroblasts (109). FSP-1 is implicated in forming functional fibroblasts by EMT by direct involvement in reshaping of cytoskeletal architecture (108). Transcriptional factors CArG box–binding factor–A (CBF-A) and KRAB-associated protein 1 (KAP-1) (110, 111) bind a proximal cis-acting response element within FTS-1 promoter, fibroblast transcription site–1 (FTS-1), and initiate transcription of the FSP-1 gene (109). The same response element has been identified in other genes associated with EMT (Twist, Snail, vimentin, collagen α1(I), and α-SMA). E-cadherin, β-catenin, and ZO-1 are negatively regulated by transcriptional factors CBF-A/KAP-1, suggesting an important synergistic role for CBF-A/KAP-1 proteins in TGF-β1–induced EMT (90, 109).
In addition to EMT, endothelial cells can also undergo endothelial-to-mesenchymal transition (EndMT) and contribute to fibrogenesis. EndMT contributed to smooth muscle–like cells in hypoxia-induced pulmonary vascular remodeling and was regulated by myocardin (112). EndMT has been also reported in cardiac fibrosis. Fibroblast like cells were originated from Tie1+ endothelial cells (4). Similar to EMT, EndMT is induced by TGF-β1 via Smad3 and antagonized by BMP-7 (4).
Although EMT and EndMT contribute to a population of collagen secreting cells, their role in fibrosis is controversial (113). Similar to that, fibrocytes and bone marrow derived myofibroblasts also contribute to ECM deposition, but to much lesser extend than tissue resident myofibroblasts.
Fibrocytes.
Fibrocytes are implicated in fibrosis in the skin, lungs, kidneys, and liver. Originally identified a decade ago by Bucala et al., they remain one of the most intriguing cells in the pathogenesis of fibrosis (114). Fibrocytes have dual characteristics of lymphoid cells and fibroblasts and are defined as collagen producing CD45+ cells (Fig. 1). Originated in the bone marrow, fibrocytes comprise a very small population (<1%) of the BM cells, which expand in response to injury and migrate in the blood to the injured organ (114, 115). The number of fibrocytes in fibrotic tissues depends on the organ and type of injury, and varies significantly, from 5% in liver to 25% in lung [Table 2; (115, 116)]. Cultured fibrocytes can differentiate into myofibroblasts in response to TGF-β1, and this effect is abrogated in the presence of serum amyloid P (117). However, it remains unclear if fibrocytes in vivo give rise to a population of α-SMA+ myofibroblasts. However, the role of fibrocytes is not limited to collagen deposition. We speculate that fibrocytes may have a unique potential of intermediary cells to mediate signals between inflammatory and fibrogenic cells. Consistent with this hypothesis, our recent findings demonstrated that fibrocytes express lymphoid markers (CD45, MHCII, MHCI), myeloid markers (CD11b, F4/80, Gr1), adhesion molecules (CD54 (ICAM-1) CD80, and CD86) and fibroblastic markers (Thy-1, collagen α1(I), see Fig. 1; Kisseleva and Brenner, unpublished observations). They also secrete growth factors and cytokines, which promote deposition of ECM in the local area of fibrosis (TGF-β1, MCP-1) (118). In response to injury fibrocytes migrate to lymphoid organs [spleen, lymph nodes (115)]. They acquire high plasticity and can differentiate into adipocytes or myofibroblasts when cultured in special conditions (115, 119). Fibrocytes may represent a common progenitor of certain myelomonocytic lineages capable of maturation into cells with myofibroblast phenotype (Kisseleva and Brenner, unpublished observations).
Fibrocytes express a number of chemokine receptors such as CCR2, CCR3, CCR5, CCR7, and CXCR4. Migration of fibrocytes to the injured organ requires chemokine/chemokine receptor interaction. Thus, CXCL12 plays a role in fibrocytes recruitment to the injured lungs in two different models of lung fibrosis (bleomycin- and FITC-induced pulmonary fibrosis), and CXCR4 or CCR2 receptors are involved in CXCL12 signaling (116, 120). However, in renal fibrosis, fibrocyte migration seems to be regulated by CCL21-CCR7 interaction and is reduced in the absence of CCR7 [CCR7-/- mice (121)]. Although the role of chemokine signaling has to be further evaluated, multiple chemokine receptors are evidently involved in direct fibrocytes recruitment to the injured organ or synergistically facilitate this process.
Fibrocytes are implicated in the pathogenesis of human diseases, especially in nephrogenic fibrosing dermopathy (122) and in murine models of human diseases. The number of infiltrating fibrocytes correlates with the intensity of fibrosis. In mice, fibrocytes infiltrate the interstitium after ureteral ligation, especially the corticomedullary regions, peaking at day 7 (121, 123). Fibrocytes may be involved in the onset of fibrosis. CD45+Col I+CXCR4+ fibrocytes are detected in bleomycin-injured lungs as early as 2 days even before collagen deposition was detected (120). Similar to renal fibrosis, the number of fibrocytes reached maximum at day 8 and correlated with an increased number of collagen producing cells. In BDL and CCL4 models of liver fibrosis, fibrocytes migrate to the injured liver, reside in portal area in a close proximity to HSCs and inflammatory cells, and constitute 5–10% of all collagen producing cells (115). Although fibrocytes do not represent the major population of collagen type I producing cells, they seem to be an obligatory component of fibrosis.
BM-Derived Fibroblasts.
BM-derived cells clearly contribute to fibrosis (10, 70, 71). However, differences in study design and terminology may cause some confusion. In addition to fibrocytes, there is another population of BM-derived fibroblasts that arise from circulating mesenchymal precursors (Kisseleva and Brenner, unpublished observation). These cells, do not express myelomonocytic markers such as CD11b, MHCII, and F4/80 and lack CD45 expression or lose it rapidly in fibrotic tissues, and are capable of differentiation into tissue fibroblasts. BM-derived fibroblasts populate irradiation-damaged lungs in mice (124). In response to bleomycin-induced injury, BM-derived fibroblasts populated lungs and contributed to lung fibrosis. These cells expressed telomerase reverse transcriptase but failed to up-regulate expression of α-SMA in vitro (125). In CCL4-injured liver, recruitment of BM-derived myofibroblasts was reported that expressed collagen, α-SMA, desmin, and vimentin (126, 127). Whether circulating mesenchymal cells develop into α-SMA+ myofibroblasts has to be determined, but they most likely represent a population different from fibrocytes.
Matrix-Induced Activation of Myofibroblasts
In addition to TGF-β1 and other fibrogenic stimuli, fibrotic changes in the cellular microenvironment can significantly affect the phenotype of tissue-resident collagen producing fibroblasts (73). Adjacent connective tissue serves as a structural scaffold that determines fibroblasts morphology. Depending on the ECM composition, fibroblasts maintain quiescence or activate into myofibroblasts. For example, in the presence of laminin, a major component of basement membranes, fibroblasts remain quiescent. In contrast, fibroblasts subjected to extracellular stress caused by abnormal ECM (e.g., fibronectin, collagen type I and III), proliferate and obtain a myofibroblastic phenotype. Similar process occurs in culture. Activated by rigid plastic material, fibroblasts secrete ECM and form stress fibers–induced cell-matrix junctions, which further facilitate ECM remodeling, suggesting that there is a feedback activation loop [reviewed in (73)]. In other words, increasing deposition of abnormal ECM accelerates activation of collagen producing cells.
The impact of ECM on liver cells has been documented (128). The state of HSCs, the major collagen producing cells in liver, is affected by direct interaction with ECM and depends on ECM content. In normal liver, quiescent HSCs are surrounded by non-electron dense basement membrane–like matrix (129). In response to injury, the composition of ECM progressively changes toward fibrillar collagen, further accelerating activation of HSCs (127). Similarly, spontaneous activation of quiescent HSCs is attenuated when cultured on basement membrane–like ECM (130). Plastic activated HSCs reverse their phenotype and down-regulate fibroblast markers if transferred to basement membrane–like ECM (130). Moreover, fibrocyte differentiation into myofibroblasts is induced in the absence of TGF-β1 when cultured on plastic or collagen type I containing matrix (Kisseleva, unpublished observations). These studies strongly suggest that matrix-myofibroblast interaction contributes to the pathogenesis of fibrosis.
Although the role of ECM in activation of pulmonary and renal fibroblasts is not well defined, studies in other tissues demonstrate similar results. Thus, lens epithelial cells proliferate and express myofibroblastic markers when cultured on collagen I-, collagen IV-, or fibronectin-, but not laminin-coated plates (131). In skin, analysis of cultured keloid fibroblasts demonstrated that not only TGFβ1 and composition of ECM regulate expression of α-SMA, but also tension in the matrix is critical for myofibroblast phenotype (132). Tension, rigidity, or stiffness of ECM is another characteristic that regulates fibroblast phenotype (133, 134). Fibroblasts cultured in a three-dimensional network of native collagen demonstrated that mechanical tension is an important mechanism underlying tissue repair and fibrosis. Mechanically stressed fibroblasts exhibit a phenotype characteristic of scleroderma fibroblasts (deposition of ECM and synthesis of fibrotic cytokines/chemokines), while fibroblasts on relaxed ECM secrete proteases and non-fibrogenic cytokines (133). In addition, culture models have demonstrated that HSC activation depends on the rigidity of the ECM and that HSC activation is accelerated by stiffness of ECM in the surrounding microenvironment (134).
Reversibility of Fibrosis
Reversibility of fibrosis has been more intensively studied in the liver than in kidneys or lungs. ECM remodeling and regression of fibrosis is always associated with withdrawal of chronic injury. Thus, in scarring of the kidney, removal of cyclosporine results in reversibility of chronic cyclosporine nephrotoxicity (135, 136). Resolution of liver fibrosis results from the withdrawal of the fibrosing insult (e.g., HBV, HCV) (19). This process correlates with decreased pro-inflammatory cytokines and TGF-β1, HSC apoptosis, and prevalence of anti-fibrogenic stimuli over fibrogenic signals. Loss of TGF-β1 signaling is critical, and the number of activated HSCs/myofibroblasts rapidly declines during resolution of liver fibrosis (71). Several mechanisms are implicated in the apoptosis of activated HSC: 1) activation of death receptor-mediated pathways (Fas or TNFR-1 receptors) and caspases 8 and 3; 2) up-regulation of pro-apoptotic proteins (e.g., p53, Bax, caspase 9); and 3) decrease of pro-survival genes (e.g., Bcl-2) (137). A population of liver associated natural killer (NK) cells and γδ T (NKT) cells stimulate apoptosis of activated HSCs. Their killing activity is enhanced in the presence of IFN-γ (138). Several studies demonstrate that activation of IL-1/iNOS-dependent pathways also facilitates HSCs apoptosis. Once the source of excessive ECM (activated HSCs) is eliminated, the collagen degradation increases (19). Increased collagenase activity is the primary pathway of fibrosis resolution. At this stage, activated macrophages/Kupffer cells secrete matrix metalloproteinases, e.g., MMP-13 interstitial collagenase, responsible for matrix degradation (139, 140). Moreover, high activity of collagen degrading enzymes correlates with decreased TIMPs, tissue inhibitors of matrix metalloproteinasesis (137). Whether cirrhosis can reverse to normal liver architecture remains controversial (127, 141). However, significant improvement in hepatic structure and function provide evidence of regression of liver fibrosis (142). However, ECM remodeling is limited in advanced fibrosis. Formation of non-reducible cross-linked collagen and an ECM rich with elastin fibers prevents its degradation. This pathophysiological state may produce irreversible cirrhosis and is considered as the “point of no return” (127, 142). Much less is known about reversibility of lung fibrosis. Loss of the integrity of the basement membrane and collapse of primary alveolar structure produce more severe damage of the lung tissue and may prevent resolution of fibrosis (143).
Concluding Remarks
Fibrosis is a complex mechanism, initiated to combat the injury and isolate the impaired tissue. However, it is important to raise an awareness that fibrogenesis does not only affect a single organ, but causes a systemic response and damage to other organs and tissues. In support to this hypothesis, administration of CCl4 affects lymphoid organs, lungs and kidneys in addition to the targeted liver. Moreover, systemic effect on other organs occurs in injured lungs and kidneys (59). Therefore, the collaborative efforts of modern interdisciplinary sciences, including multiple organs biology, pathology, computational biology, immunology, and transgenic technologies will significantly advance our understanding of fibrosis.
Progression of Fibrosis a
Origin of Collagen Producing Cells in Fibrosing Organs a
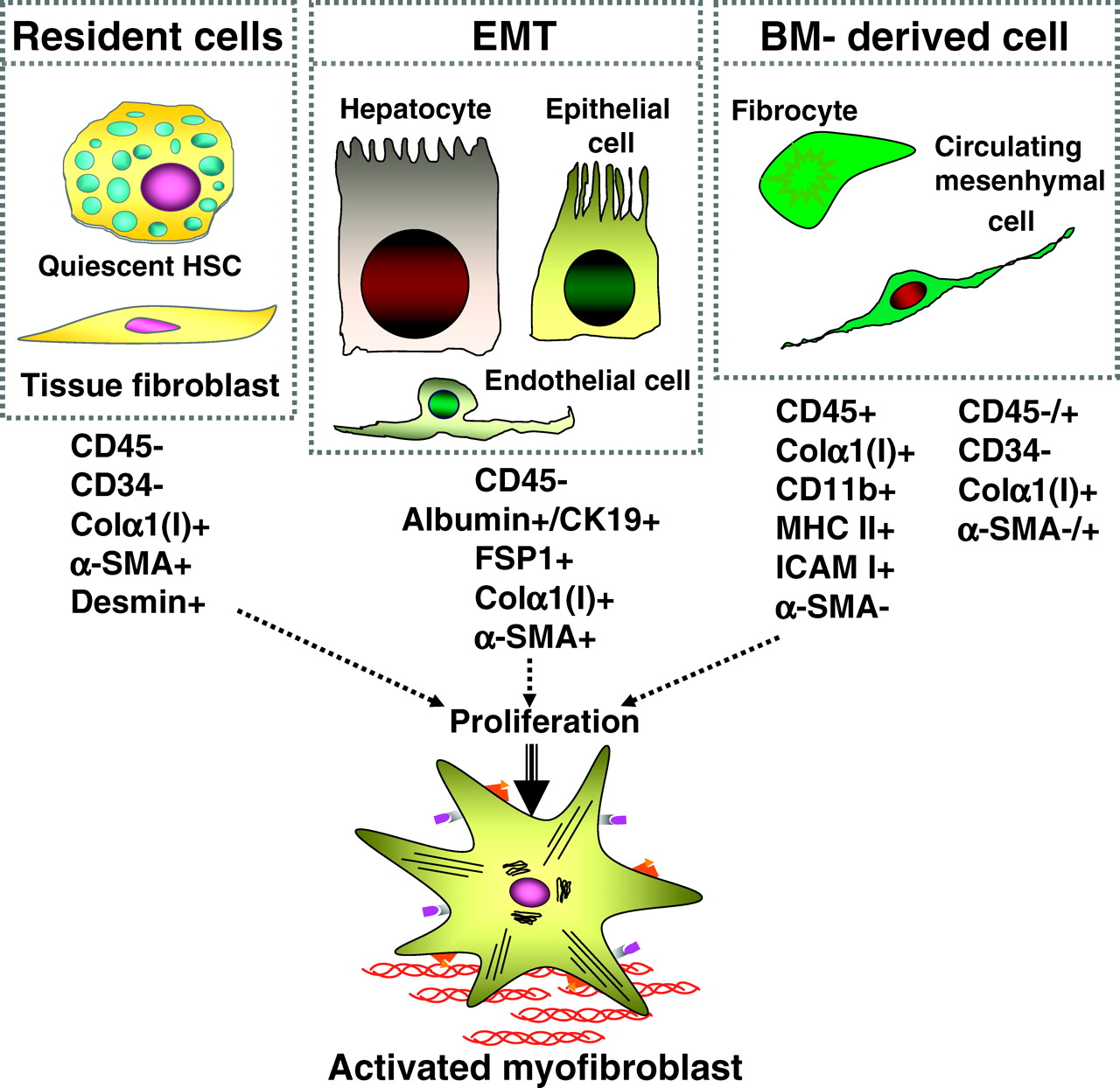
Origin of collagen producing cells. There are three general sources of fibrogenic cells: endogenous fibroblasts or fibroblast-like cells, epithelial to mesenchymal transition, and recruitment of fibrocytes from the bone marrow. Explanation is in the text. HSCs, hepatic stellate cells; α-SMA, α-smooth muscle actin; Colα1(I), collagen α1(I); EMT, endothelial to mesenchymal transition; FSP1, fibroblast-specific protein 1. A color version of the figure is available in the online version of the journal.
Footnotes
This work was supported by a Liver Scholar Award from the American Liver Foundation.