
Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) have been shown to exhibit potent anticancer effects in vitro and in vivo. One of the mechanisms by which NSAIDs suppress tumorigenesis is inhibition of angiogenesis and metastasis. In this study, we used a microarray system to study the change of expression profile of metastasis-related genes regulated by NS398, a NSAID and a cyclooxygenase-2 (COX-2) inhibitor. We found that several negative regulators of cell invasion, including secreted protein acidic and rich in cysteine (SPARC), thrombospondin 1 (TSP-1), thrombospondin 3 (TSP-3), and tissue inhibitors of matrix metalloproteinase-2 (TIMP-2) are upregulated by NS398. In addition, we demonstrated that upregulation of SPARC expression by NS398 in human lung cancer cells is mediated by promoter demethylation and associated with a decrease in DNA methyltransferase (DNMT) expression. This is the first report to show that NS398 can inhibit the expression of DNMT1 and 3b. Functional assay indicated that SPARC is a critical mediator for NS398 to inhibit cell invasion. Our results provide new insights for the understanding of the anticancer actions of NSAIDs.
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) have been shown to exhibit anticancer and chemopreventive effects on various types of human cancer (1, 2). The anticancer actions of NSAIDs are thought to be mediated by several mechanisms. First, NSAIDs can inhibit proliferation of cancer cells. Recent studies indicate that the progression of cell cycle is governed by cyclins and cyclin-dependent kinases (CDKs; Ref. 3). The progression of cell cycle from G1 to S phase is controlled by D- and E-type cyclins and their major catalytic subunits CDK4, 6, and 2 (4, 5). The progression through S, G2, and M is mainly regulated by A- and B-type cyclins and CDK2 (6). Conversely, a group of negative regulatory proteins may inhibit cell-cycle control by repressing the enzymatic activity of the cyclin/CDK complexes. These proteins, termed CDK inhibitors (CDKIs), are classified into two main groups. The first class is the INK4 family (including p15INK4B, p16INK4A, p18INK4C, and p19INK4D), which inhibits specifically the activity of cyclin D–associated CDK4 and 6 (7). The second class is the CIP/KIP family (including p21WAF1, p27KIP1, and p57KIP2), which has broad specificity and inhibits the activity of most cyclin-CDK complexes (8, 9). NSAIDs may upregulate the expression of p21WAF1 and p27KIP1 to inhibit the cyclin-CDK activity and induce growth arrest of cancer cells (10–12). Second, some NSAIDs exhibit apoptosis-inducing activity and may trigger apoptotic cell death by modulating the expression of Bcl-2 gene family or activating the death receptor–associated signaling pathways (13, 14). Third, NSAIDs may suppress tumor angiogenesis and metastasis. Previous studies demonstrated that NSAIDs repress the expression of angiogenic factors, including vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), which results in reduction of tumor angiogenesis (15, 16). Our recent studies also showed that NSAIDs effectively suppress matrix metalloproteinase-2 (MMP-2) expression and reduce invasiveness of human lung cancer cells (17, 18).
SPARC, also named as osteonectin/BM-40, is a glycoprotein that plays a critical role in the control of cell-matrix interaction and is involved in various physiologic processes, including tissue remodeling, cell adhesion, migration, angiogenesis, and tumor metastasis (19–21). SPARC may positively or negatively regulate tumor progression. Previous studies demonstrated that SPARC is overexpressed in human cancers and is associated with tumor angiogenesis and invasion (20, 21). Conversely, SPARC has been shown to inhibit angiogenesis and growth of Lewis lung cancer, neuroblastoma, and ovarian cancer cells (22, 23). Therefore, SPARC has multiple functions and may exhibit cell type–specific actions.
To further identify the important mediators for NSAID-induced inhibition of tumor angiogenesis and metastasis, we used the Human Extracellular Matrix and Adhesion Molecules Gene Array system to study the change of expression profile induced by a NSAID and COX-2 inhibitor NS398. NS398 (N-[2-cyclohexyloxy-4-nitrophenyl] methanesulfonamide), which exhibited analgesic and antipyretic effects, was originally synthesized by Futaki et al. (24, 25). Subsequent study demonstrated that this drug selectively inhibited COX-2 and produced much fewer gastrointestinal lesions in rats (26). We had tested the effect of NS398 on human lung cancer cells and found that it inhibited cell proliferation via induction of an important CDKI p27KIP1 to induce G1 growth arrest (27, 28). We also found that NS398 attenuated the invasiveness of lung cancer cells via downregulation of MMP-2 (17, 18). In this study, we found that an antimetastatic gene, SPARC, was increased by NS398. Our real time polymerase chain reaction (PCR) and Western blotting data confirmed the microarray results that NS398 upregulated SPARC mRNA and protein level in a dose-dependent manner. Because DNA methylation catalyzed by DNA methyltransferases (DNMTs) including DNMT-1, 2, 3a, 3b, and 3L is a crucial epigenetic modification and is associated with the silencing of SPARC in human cancers (29, 30), we performed methylation-specific PCR (MSP) to investigate the methylation status of SPARC promoter. Our results showed that upregulation of SPARC by NS398 in human lung cancer cells is mediated by promoter demethylation and associated with a decrease in DNMT expression.
Materials and Methods
Cell Culture and Reagents.
A549 lung cancer cells were cultured in Dulbecco’s modified Eagle’s medium and F12 nutrition mixture (DMEM/F12) supplemented with 10% heat-inactivated fetal calf serum (FCS) and antibiotics. NS398, indomethacin, and sulindac sulfide were obtained from Sigma (St. Louis, MO). The monoclonal antibody (clone ON1–1, catalog No. 33–5500) against SPARC was obtained from Zymed Laboratories (S. San Francisco, CA). The isotype of this antibody is IgG1, and it was obtained by fusing the mouse myeloma cell line P3U1 with the spleen cells of BALB/c mouse after immunization with bovine SPARC. This antibody specifically reacts with human and bovine SPARC.
Microarray and Data Analysis.
Total RNA extraction was prepared using the RNeasy Kit (Qiagen, Hilden, Germany) according to the instructions of the manufacturer. The integrity of RNA was checked by agarose gel electrophoresis. Expression of metastasis-associated genes was investigated by using SuperArray microarray GEArray Q series (Human Extracellular Matrix & Adhesion Molecules gene Array) system (SuperArray Bioscience Corporation, Frederick, MD). Isolated RNA (0.5 μg) was used as a template for biotin-labeled cDNA probe synthesis, through a MMLV reverse transcriptase reaction. Biotin-labeled probes were used to hybridize with a GEArray HS-010 membrane. After hybridization, the membrane was incubated with streptavidin-alkaline phosphatase conjugate. The signals were developed with CDP-Star substrate and detected on x-ray films. The autoradiograms were analyzed by using GEArray Analyzer, available on SuperArray’s website (www.superarray.com). The signal intensities of target genes were normalized to the signals of actin or glycer-aldehyde-3-phosphate dehydrogenase (GAPDH) on the same microarray blot. Similar results were obtained by using either one for normalization.
Western Blot Analysis.
Cells were treated with vehicle (0.1% DMSO), NS398 (100 μM), indomethacin (100 μM), or sulindac (5 μM) in 10% FCS medium for 24 hrs. Conditioned medium was collected, and cell number was determined by using the hemocytometer. Conditioned medium from an equal number of cells was separated by SDS-polyacrylamide gel electrophoresis, and Western blot analysis was performed to detect the SPARC protein in the culture medium.
RNA Extraction and Real-time Reverse Transcription (RT)-PCR.
Cells were treated with vehicle or NS398 for 24 hrs. Total RNA was isolated from cells by the RNeasy Kit, and 1 μg of RNA was reverse-transcribed into complementary DNA. Expressions of various target genes were quantified by real-time RT-PCR using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA). GAPDH was served as an internal control for total complementary DNA content. One nanogram of cDNA was amplified by using the MJ Mini real-time PCR detection system (Bio-Rad). The PCR primers used are: SPARC-forward, 5′-GTGCAGAG GAAACCGAAGAG-3′; SPARC-reverse, 5′-TGT TTGCAGTGGTGGTTCTG-3′; TSP-1-forward, 5′-ACTGTCCCTATGTGCCCAATGC-3′; TSP-1-reverse, 5′-CCATCGCCGTCACAGGAGTCCTTC-3′; TSP-3-forward, 5′-TTTCACATCTGCCAGTCAGG-3′; TSP-3-reverse, 5′-AGCTACCATCTGCCGAGACT-3′; TIMP-2-forward, 5′-GAAGAGCCTGAACCACAGGT-3′; TIMP-2-reverse, 5′-CGGGGAGGAGATGTAGCAC-3′; DNMT1-forward, 5′-GATGTGGCGTCTGTGAGGT-3′; DNMT1-reverse, 5′-CCTTGCAGGCTTTACATTTCC-3′; DNMT3b-forward, 5′-CAAATGGCTTCAGATGTTGC-3′; DNMT3b-reverse, 5′-TCCTGCCACAAGACAAACAG-3′; GAPDH-forward, 5′-AAGGCTGGGGCTCATTTGC-3′; and GAPDH-reverse, 5′-GCTGATGATCTTGAGGCT GTTG-3′. The PCR conditions are denaturation at 95°C for 15 secs, annealing at 61°C for 30 secs, and extension at 72°C for 30 secs. The PCR reaction was run for 40 cycles. Experiments were done in triplicate, and results from three independent experiments were averaged.
Invasion Assay.
In vitro invasion assay was performed by using 24-well transwell units with polycarbonate filters (pore size 8 μm) coated on the upper side with Matrigel (Becton Dickinson Labware, Bedford, MA). Cells were collected and 5000 cells in 100 μl of serum-free medium containing 0.1% DMSO or 100 μM of NS398 and 5 μg/ml nonimmune mouse immunoglobulin or anti-SPARC antibody were placed in the upper part of the transwell unit and allowed to invade for 24 hrs. The lower part of the transwell unit was filled with 10% FCS medium. After incubation, noninvaded cells on the upper part of the membrane were removed with a cotton swab. Invaded cells on the bottom surface of the membrane were fixed in formaldehyde, stained with Giemsa solution, and counted under a microscope. The invaded cell number of cells cultured without NS398 and antibody was defined as 100%. Results from three independent experiments were expressed as mean ± SD. Nonimmune mouse gamma immunoglobulin was purchased from Jackson Immunoresearch Laboratories (West Grove, PA). Anti-SPARC antibody with the isotype of IgG1 was obtained by fusing the mouse myeloma cell line P3U1 with the spleen cells of BALB/c mouse after immunization with bovine SPARC and was purchased from Zymed Laboratories.
Methylation-Specific Polymerase Chain Reaction (MSP).
Genomic DNA was isolated from cells treated without NSAIDs or with sulindac sulfide (SS, 20 μM), NS398 (NS, 100 μM), indomethacin (Indo, 100 μM) or 5′-azacytidine (AZC, 5 μM) for 24 hrs by using the QIAamp DNA Mini kit (Qiagene). The DNA was modified with sodium bisulfite and analyzed according to the procedures of CpGenome DNA Modification kit (Chemicon). The primers used to amplify the unmethylated (U) SPARC promoter are U-forward: 5′-TTTTTTAGATTGTTTGGAG AGTG-3′ and U-reverse: 5′-AACTAACAACATAAAC AAAAATATC-3′. The primers used for amplification of methylated SPARC promoter are M-forward: 5′-GAGAGCGCGTTTTGTTTGTC-3′ and M-reverse: 5′-AACGACGTAAACGAAAATATCG-3′. PCR was carried out in a thermal cycle for 36 cycles (denaturation at 95°C for 1 min, annealing at 60°C for 2 mins, and extension at 72°C for 2 mins) followed by a final 5-min extension at 72°C. PCR products were separated in 3% agarose gels, stained with ethidium bromide, and visualized under ultraviolet illumination.
Statistical Analysis.
Student’s t test was used to evaluate the difference between various experimental groups. Differences were considered to be significant at P < 0.05.
Results and Discussion
To identify the important mediators for NSAID-induced inhibition of tumor angiogenesis and metastasis, we used the Human Extracellular Matrix and Adhesion Molecules Gene Array system, which contained 96 metastasis-associated genes to study the change of expression profile induced by a NSAID and COX-2 inhibitor NS398. We chose this drug for assay because our previous studies have demonstrated that NS398 might inhibit cell proliferation via induction of p27KIP1 in human lung cancer cells (27, 28). In addition, we also found that this drug exerts potent anti-invasive activity on lung cancer cells (17, 18). More importantly, NS398 had been shown to prevent the tobacco-specific carcinogenic nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis in vivo (31). As demonstrated in Table 1, several antimetastatic genes, including SPARC, TSP-1, TSP-3, and TIMP-2 were consistently increased by NS398 in two independent microarray assays. To verify the results of microarray analysis, the effect of NS398 on gene expression was investigated by quantitative RT-PCR. We found the results of quantitative RT-PCR are consistent with the data of microarray and NS398 upregulating the expression of SPARC, TSP-1, TSP-3, and TIMP-2 by 2.32-, 3.29-, 5.26-, and 1.74-fold, respectively (Fig. 1). We next focus on the regulation of SPARC by NS398. Quantitative RT-PCR results demonstrated that NS398 upregulated SPARC mRNA in a dose-dependent manner (from 1.4- to 2.2-fold, Fig. 2A). Western blot analysis also confirmed the upregulation of SPARC protein (Fig. 2B). In addition, the amount of SPARC protein in the conditioned medium was also increased (Fig. 2C). This is not a NS398-specific effect because another two NSAIDs, sulindac sulfide and indomethacin, also upregulated SPARC expression by 1.8- and 2.2-fold, respectively (Fig. 1D). We next studied the involvement of SPARC in NSAID-induced inhibition of cell invasion. Addition of NS398 potently suppressed the invasive ability of A549 cells (Fig. 3). When cells were incubated with anti-SPARC antibody, we found that inhibition of invasion by NS398 was partly reversed. On the contrary, addition of nonimmune mouse gamma immunoglobulin (with the same isotype to anti-SPARC antibody) had no effect. These results suggest that SPARC is one of the mediators for NS398 to suppress cell invasion.
How NSAIDs regulate gene expression is an important issue for the understanding of the anticancer actions of these drugs. The molecular mechanisms by which NSAIDs inhibit gene expression have been characterized. First, NSAIDs may modulate the mitogen-activated protein kinase (MAPK) signaling pathway to inhibit gene expression. For example, NSAIDs repressed the p38 MAPK activity to inhibit the activation of NF-AT transcription factor and reduce the expression of COX-1 and COX-2 (32). We also found that NSAIDs inhibit the expression of MMP-2 by suppressing extracellular signal-regulated kinase (ERK) signaling pathway (18). Second, NSAIDs may inhibit IKK activity and suppress NF-κB–mediated gene activation. For instance, a NSAID sulindac has been shown to block the NF-κB–mediated survival signals through inhibition of IKK-beta (33). In addition, NSAIDs also repress NF-κB–induced expression of COX-2 and cyclin D1 (34). However, how NSAIDs upregulate gene expression is poorly understood. A recent study demonstrated that NSAIDs upregulated the expression of TSP-1 to repress tumor growth (35). The authors showed that this induction is mediated via the transcription factor Erg-1. Interestingly, another study came from the same group also demonstrated that COX-2 inhibitors stimulated Erg-1 to activate a tumor suppressor, nonsteroidal anti-inflammatory drug-activated gene (NAG-1) in cancer cells to inhibit tumorigenesis (36). Therefore, Erg-1 is one of the mediators for the NSAID-induced gene expression and tumor suppression. However, whether NSAIDs stimulate SPARC via the same mechanism is questionable. Recent studies indicated that down-regulation of SPARC expression in cancer cells is mainly caused by promoter hypermethylation. Ito et al. showed that reduction of SPARC is associated with hypermethylation of gene promoter in human lung cancer (37). Similarly, Suzuki et al. found that loss of SPRAC expression was observed in 83% of non–small-cell lung cancer cell lines and was strongly correlated with promoter methylation (38). In addition, SPARC promoter methylation was detected in 69% of lung tumor tissues and overall survival of adenocarcinoma was poorer in patients with SPARC promoter methylation.
DNA methylation is a crucial epigenetic modification and is frequently associated with the silencing of tumor suppressor genes in cancers (29). Methylation of the CpG sites on DNA is catalyzed by DNMTs, and five DNMTs have been identified in mammalian cells (30). DNMT1 is a maintenance methyltransferase that copies pre-existing methylation pattern onto the new strand of replicated DNA. DNMT3a and 3b are responsible for introducing cytosine methylation at previously unmethylated CpG sites. DNMT2 and 3L exhibit very weak or no intrinsic DNA methyltransferase activity, and their biologic functions are still unclear. Because SPARC promoter is frequently methylated in lung cancer cells, we tested whether NSAIDs may induce promoter demethylation of SPARC gene in A549 cells. As shown in Figure. 4A, NS398, sulindac sulfide, and indomethacin all reduced promoter methylation of SPARC gene. This is not a nonspecific effect because 5′-azacytidine, a DNMT inhibitor also induced promoter demethylation under the same experimental condition (Fig. 4A). In addition, 5′-azacytidine also increased SPARC expression by fourfold in A549 cells (data not shown). We next tested the possibility whether NSAIDs may directly inhibit DNMT expression. Our results showed that the expression of DNMT3a was very low in A549 cells (data not shown). DNMT1 and DNMT3b were expressed in A549 cells, and NS398 suppressed their expressions by 37% and 62%, respectively (Fig. 4B).
To the best of our knowledge, this is the first report to demonstrate that NSAIDs may suppress the expression of DNMTs. Because epigenetic silencing of tumor suppressor genes by promoter hypermethylation is frequently found in human cancers, DNMT inhibitors are thought to be a novel class of anticancer drugs because they may induce promoter demethylation and restore the expression of tumor suppressor genes in cancer cells. Indeed, some nucleoside or nonnucleoside DNMT inhibitors are progressing into clinical trials. For example, two DNMT inhibitors 5′-azacytidine and decitabine have been tested in phase I and II clinical trials for the treatment of different cancers (39). More recently, 5′-azacytidine (also named as Vidaza) has been approved for the treatment of myelodysplastic syndrome, a preleukemic bone marrow disorder (39). Although NSAIDs do not directly inhibit the enzymatic activity of DNMTs, these drugs may repress DNMT expression and may be developed as a new class of demethylating agents.
Recently, a number of studies suggest that NSAIDs are effective adjuvant drugs for chemotherapy and radiotherapy. These conclusions were initiated by the observation that NSAIDs enhanced growth inhibition in combination with a chemotherapeutic drug gemcitabine in pancreatic carcinoma cells (40). A recent clinical trail also demonstrated that celecoxib enhanced the response of paclitaxel and carbo-platin in early-stage non–small-cell lung cancer (41). Additionally, Kishi et al. reported the preferential enhancement of tumor radioresponse by a COX-2 inhibitor SC-236 (42). Subsequent investigations strongly suggested that NSAIDs or COX-2 inhibitors are radiation sensitizers and may effectively improve the response of cancer cells to radiotherapy (43). However, the molecular mechanism by which NSAIDS enhance the anticancer action of chemotherapeutic drugs or radiation is not clear. Previous studies demonstrated that a number of proapototic genes, including death-associated protein kinase (DAPK), caspase-8, and target of methylation-induced silencing 1 (TMS1) are silenced by promoter methylation in cancer cells (44, 45). It is possible that NSAIDs may reverse the promoter methylation of these target genes to enhance the effect of chemotherapy or radiotherapy. This hypothesis is supported by the observation that NSAIDs reactivate the expression of some hypermethylated genes in A549 lung cancer cells (data not shown). Taken together, this study demonstrates that NSAIDs may induce promoter demethylation and reactivate expression of some metastasis suppressor genes in lung cancer cells. These results also provide new insights for the anti-cancer actions of NSAIDs.
Typical Upregulated Genes in NS398-Treated A549 Cancer Cells
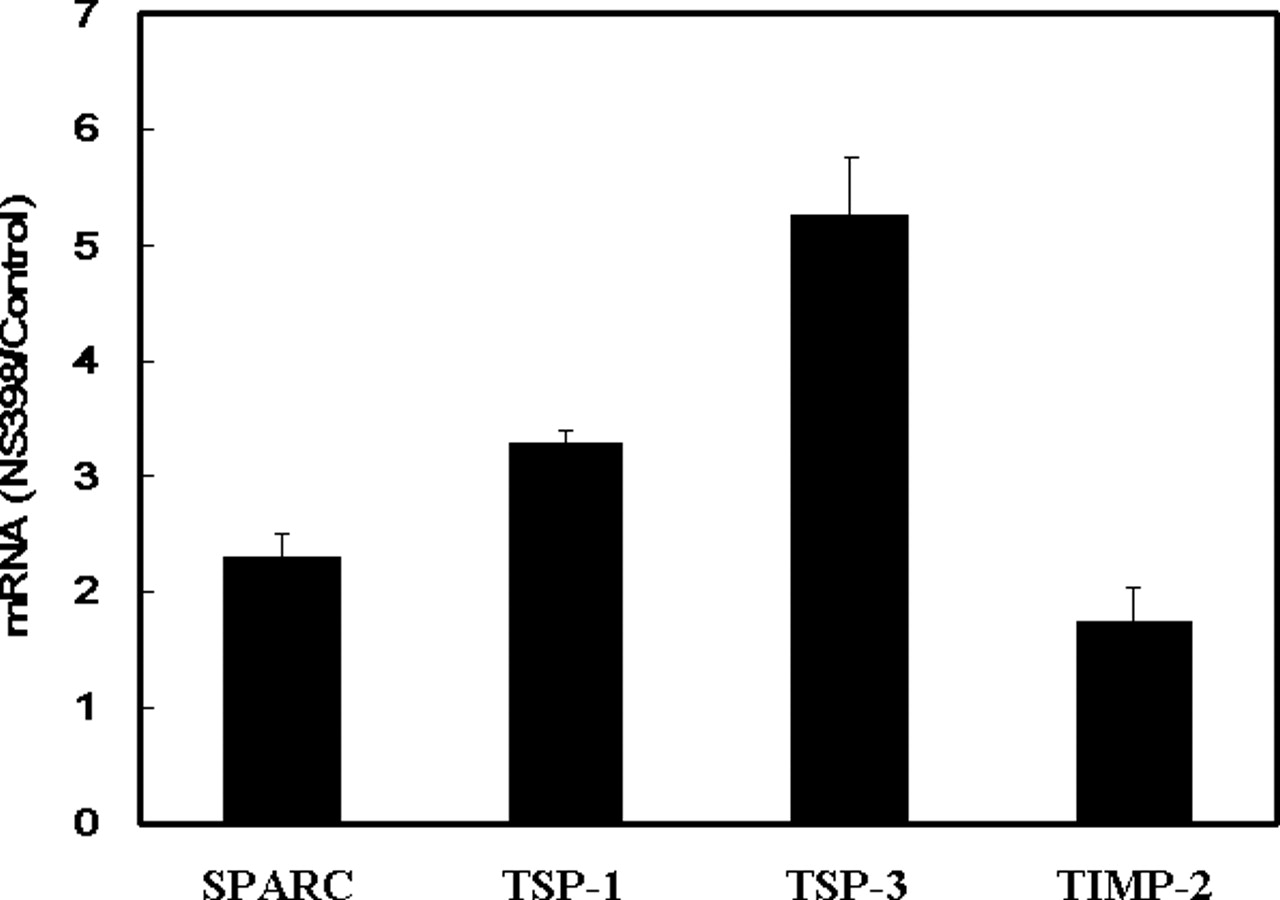
NS398 upregulates antimetastatic genes in A549 human lung cancer cells. Cells were treated with vehicle (0.1% DMSO) or 100 μM of NS398 for 24 hrs. Total RNA extraction and quantitative RT-PCR was performed to investigate the mRNA level of SPARC, TSP-1, TSP-3, and TIMP-2. The mRNA level of target genes was normalized by GAPDH and the ratio of NS398- and vehicle-treated cells (NS398/Control) from three independent experiments was expressed as mean ± SD.

Upregulation of SPARC by NS398. (A) Cells were treated with various concentrations of NS398 for 24 hrs. Quantitative RT-PCR was performed to investigate the mRNA level of SPARC. The SPARC mRNA level was normalized by GAPDH and the ratio of NS398- and vehicle-treated cells (NS398/Control) from three independent experiments was expressed as mean ± SD. (B) Cells were treated with various concentrations of NS398 for 24 hrs, and cellular proteins were harvested for analysis. Western blotting was performed to investigate the SPARC protein level, and actin was used as an internal loading control. (C) Conditioned medium of cells treated with 100 μM of NS398 was harvested at 24 hrs after treatment and normalized by adhesive viable cell number. Samples were subjected to SDS-polyacrylamide gel electrophoresis, and the amount of SPARC protein in the conditioned medium was detected by Western blotting. (D) Cells were treated with vehicle, sulindac sulfide (SS, 20 μM) or indomethacin (Indo, 100 μM) for 24 hrs. Quantitative RT-PCR was performed to investigate the mRNA level of SPARC. The SPARC mRNA level was normalized by GAPDH and the ratio of drug- and vehicle-treated cells (Drug/Control) from three independent experiments was expressed as mean ± SD.
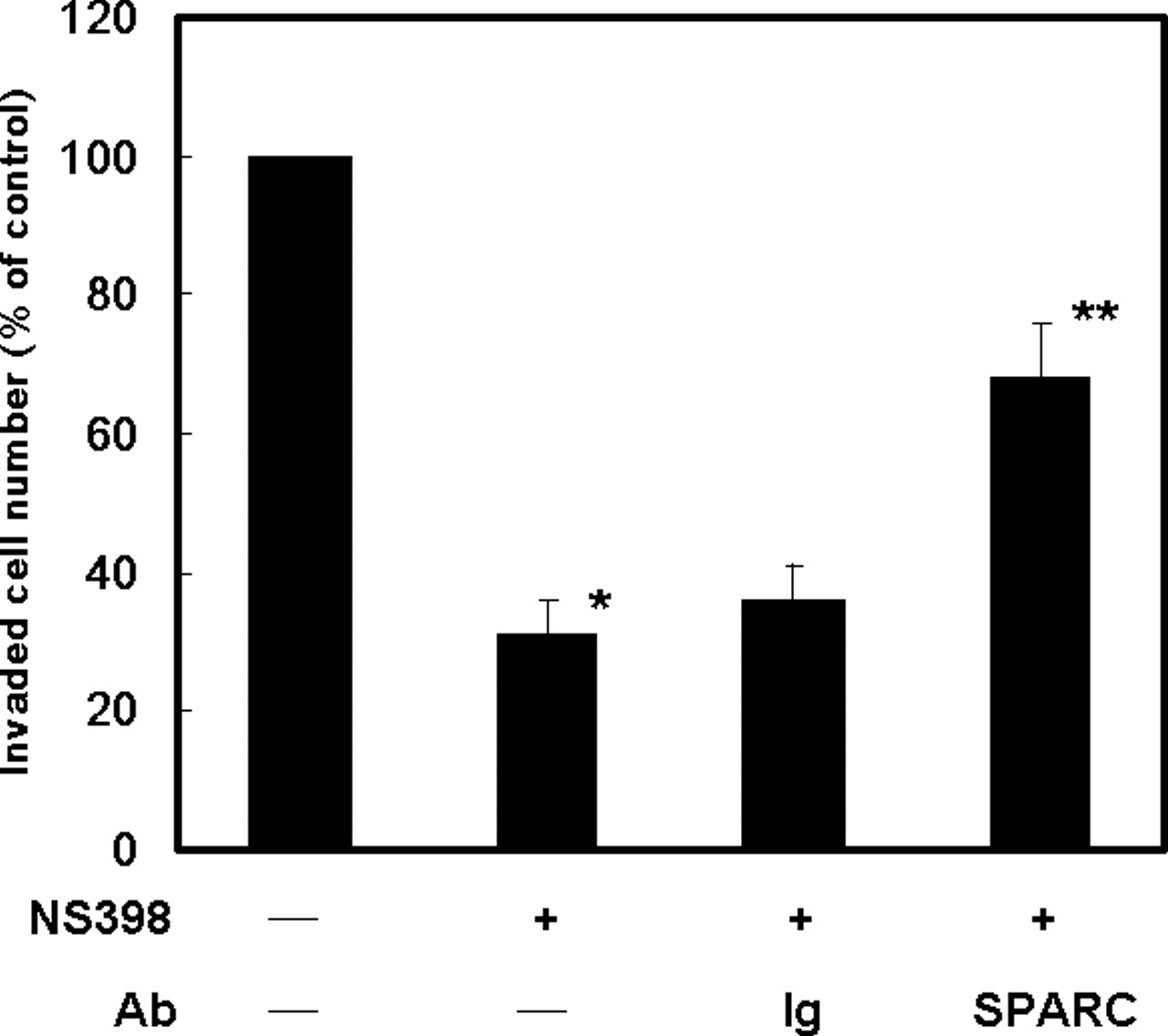
SPARC is a mediator for NS398 to inhibit cell invasion. In vitro invasion assay was performed by using 24-well transwell units with polycarbonate filters (pore size 8 μm) coated on the upper side with Matrigel. Cells were collected and 5 × 103 cells in 100 μl of medium containing 0.1% DMSO (−) or 100 μM of NS398 (+) and nonimmune mouse gamma immunoglobulin (Ig, 5 μg/ml) or anti-SPARC antibody (SPARC, 5 μg/ml) were placed in the upper part of the transwell unit and allowed to invade for 24 hrs. The lower part of the transwell unit was filled with 10% FCS medium. The invaded cell number of cells cultured without NS398 (−) and antibody (−) was defined as 100%. Results from three independent experiments were expressed as mean ± SD. *P < 0.05, when control and NS398-treated groups were compared. **P < 0.05, when the group cotreated with NS398 and anti-SPARC antibody was compared with the group treated with NS398 alone.
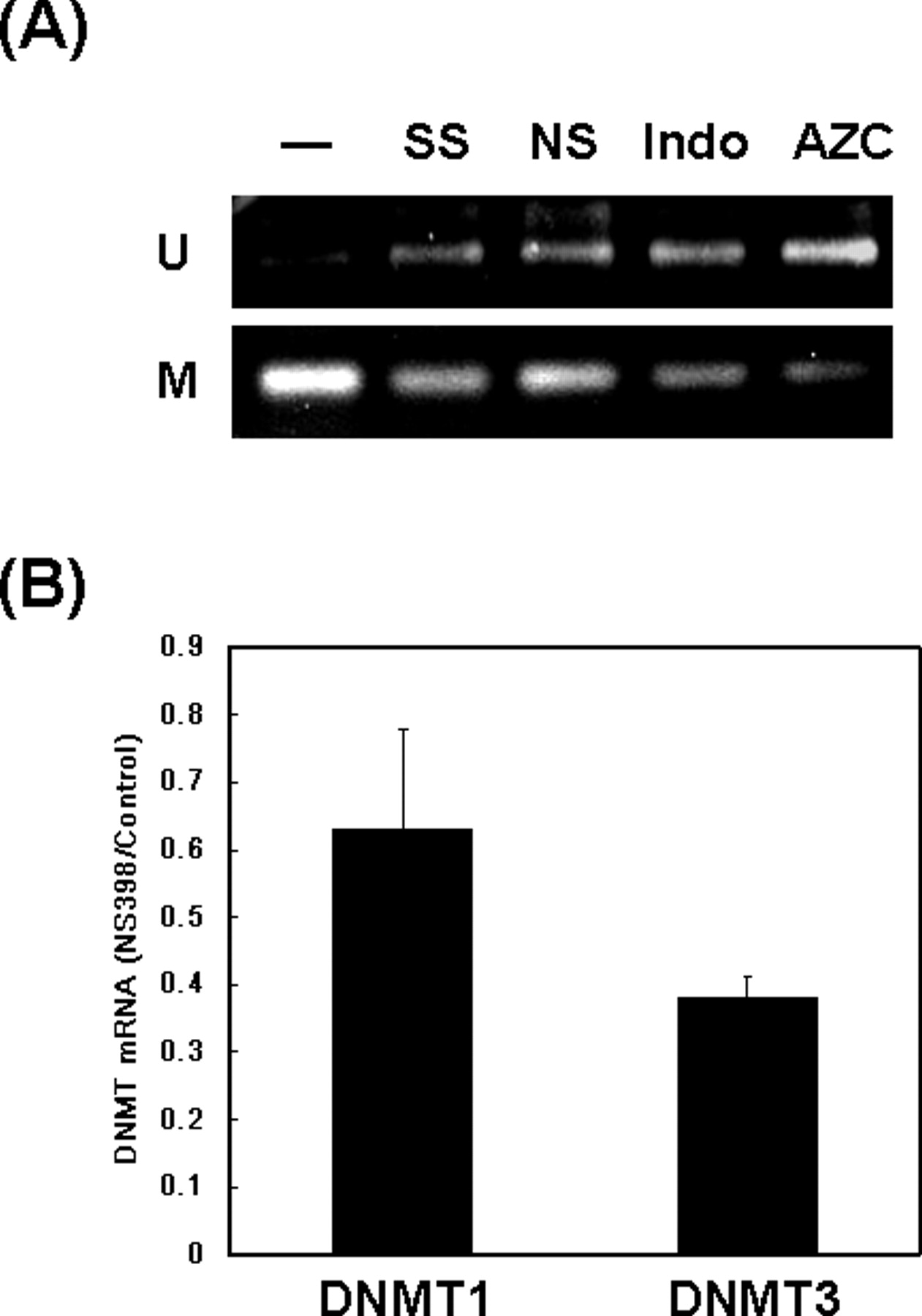
NS398 downregulates the expression of DNMTs and induces promoter demethylation of SPARC gene. (A) Genomic DNA was isolated from A549 cells treated without (−) or with sulindac sulfide (SS, 20 μM), NS398 (NS, 100 μM), indomethacin (Indo, 100 μM), or 5′-azacytidine (AZC, 5 μM) for 24 hrs. Sodium bisulfite modification and methylation-specific polymerase chain reaction (MSP) was performed as described in Materials and Methods. (B) Cells were treated with vehicle or NS398 (100 μM) for 24 hrs. Total RNA extraction and quantitative RT-PCR was performed to investigate the mRNA level of DNMT1 and DNMT3b. The DNMT mRNA level was normalized by GAPDH and the ratio of NS398- and vehicle-treated cells (NS398/Control) from three independent experiments was expressed as mean ± SD.
Footnotes
This study was supported by the grants from Center for Gene Regulation and Signal Transduction Research, National Cheng Kung University and National Sun Yat-Sen University-Kaohsiung Medical University Joint Research Center.