Abstract
Saponins are amphiphilic substances consisting of a hydrophobic backbone with one or two hydrophilic sugar units. Recently it was shown that saponinum album (SA) from Gypsophila paniculata L. enhanced cytotoxicity of a saporin-based chimeric toxin (up to 385,000-fold) as well as the toxicity of saporin (up to 100,000-fold) with N-glycosidase activity. Previously we have shown that toxicity of other N-glycosidases such as ricin A-chain, nigrin b, and toxins such as diphtheria toxin or microcystin-LR was not enhanced by SA. This points to a specific SA-dependent mechanism of toxicity enhancement on saporin and saporin-based toxins. Although the cytotoxicity enhancing effect was observed in up to 10 different cell lines, nothing is known about the kinetic of SA under cell culture conditions. Therefore SA was titrated, and the uptake respective liberation profile of SA was investigated in ECV-304 cells. Treatment of cells with [3H]-SA leads to an immediate uptake of saponin molecules. After cells were saturated with [3H]-SA, a first equilibrium (first eq.) was reached. The first eq. was disturbed by washing until a second equilibrium was reached between the activity observed within cells and that seen in the supernatant. After a further extensive washing, a small portion of saponin molecules remained durable associated with the cell. This portion was sufficient to induce a drastic toxicity enhancement on saporin indicating a long-lasting sensitization of cells against the toxin.
Introduction
Saponins are glycosides, consisting of sugar moieties that are linked to a triterpene or steroid backbone. Due to this structural composition, saponins are surface active compounds that give stable foams when interacting with water and were shown to associate with the plasma membrane of cells and model membranes by interacting with cholesterol (1–3). Recently it has been shown that saponinum album (SA) from Gypsophila paniculata L. enhances the cytotoxicity of the N-glycosidase saporin (up to 100,000-fold) (4) as well as enhancing the toxicity of a chimeric toxin composed of saporin and human epidermal growth factor (EGF) up to 385,000-fold (5, 6). The SA-mediated enhancement of toxicity on these toxins was shown in a panel of 10 different cell lines (5–7). This indicates that there is a common SA-dependent mechanism of action, leading to shared sensitivity against saporin and other related toxins. A further group of saponins excluding SA showed no cytotoxicity-enhancing effect (5–8). The toxicity of other N-glycosidases such as the ricin A-chain, nigrin b, and toxins such as diphtheria toxin or microcystin-LR was not enhanced by SA (4). This points to a specific SA-dependent mechanism of toxicity enhancement on saporin and saporin-based toxins.
Saponins derived from SA are characterized by a formyl-function attached at position 4 of the C30 backbone (aglycon), an acidic sugar chain bound at position 3, and a second sugar chain in position 28 as shown in Figure 1 (9–11). Note the amphiphilic structure of the saponin consisting of a hydrophobic aglycone with two hydrophilic sugar units attached at carbon positions 3 and 28 of the aglycone. Although the cytotoxicity-enhancing effect on these toxins is documented in several cell lines, nothing is known about the uptake and kinetic profile of SA under cell culture conditions. We attached a low molecular-weight radio-labelled tag to SA and performed kinetic experiments in ECV-304 cells. To correlate the results of these experiments with the SA-dependent toxicity enhancement on saporin, additional cytotoxicity experiments with cold SA and saporin were performed.
Materials and Methods
[3H]-acetic anhydride (25 mCi) was obtained from Amersham (Munich, Germany). Pyridine, toluol, and saponinum album were purchased from Merck (Darmstadt, Germany). Optiphase Supermix scintillation cocktail and soluene 350 were purchased from Perkin Elmer (Wiesba-den, Germany). The tetrazolium salt 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) and phenazine methosulfate (PMS) were purchased from Sigma-Aldrich (Steinheim, Germany).
Cell Culture.
ECV-304 cells (DSMZ-No. ACC 304) were purchased from the German Cell Culture Collection (Braunschweig, Germany). Cells were cultured in modified Eagle’s medium (MEM) without phenol red supplemented with 10% fetal bovine serum (FBS).
Radio Labelling with [3H]-Acetic Anhydride.
Because labelling with acetic anhydride and pyridine (1:1) can be easily performed for sugars and saponin, we used 3[H]-acetic anhydride/pyridine to label SA.
A mixture of 2 ml pyridine, 1.75 ml cold acetic anhydride, and 0.25 ml [3H]-acetic anhydride (25 mCi) was added to 100 mg SA, and the whole mixture was swirled vigorously for 3.5 min. Aqua dest. (5 ml) was added, and the solution was cooled to room temperature. After extraction of the acetylated saponins with chloroform (3 times, each 5 ml), the organic phase (15 ml) was cleared by dropwise addition of methanol. After drying over sodium sulphate, the chloroform and methanol fraction was evaporated at 100 mbar, 40°C. Pyridine was removed by azeotrop distillation with toluol at 50 mbar, 40°C, creating a solid [3H]-SA product (8 mg). Specific, vehicle-free activity was determined by liquid scintillation and was calculated to be approximately 6 × 105 Bq/mg [3H]-SA.
Before using [3H]-acetic anhydride, the labelling method was optimized with cold acetic anhydride/pyridine. Success of the labelling of SA was verified by high-performance thin layer chromatography (HPTLC) with silica gel 60-glass plates. Eluent was the upper phase of the acetic acid (99%):aqua dest.:butanol-mixture (10:40:50). Labelling was further scrutinized by HPLC of the labelled and unmodified SA and was performed as described elsewhere (12). HPLC fractions were analysed by ESI-TOF mass spectrometry. Mass shifts of unlabelled SA were examined, and the labelled SA showed strong toxicity-enhancing properties on saporin (data not shown).
Liquid Scintillation Measurement.
For measuring the activity of tritium, a Wallac MicroBeta Trilux beta counter and Optiphase Supermix scintillation cocktail were used.
Time Course Experiments with [3H]-SA.
1 × 104 ECV-304 cells were seeded in 24-well plates and grown in 400 μL MEM to confluence. Cells were washed with 300 μl PBS to which 300 μl MEM was added. Thereafter cells were incubated with [3H]-SA (10 μg/ml) for different times, ranging from 2.5 to 240 min. After aspiration the medium, cells were washed with PBS (300 μl), trypsinized (200 μl), and then transferred to a 2 ml eppendorf tube that contained 100 μl soluene 350. This mixture (300 μl) was homogenized, and 1.7 ml Optiphase Supermix scintillation cocktail was added to each tube.
Kinetic of [3H]-SA in ECV-304 Cells.
1×105 ECV-304 cells were seeded in 6-well plates and grown in 1 ml MEM to confluence. [3H]-SA (final concentration 10 μg/ml) was added to each well, and cells were incubated for 60 min. Cells were washed (2 ml PBS) and covered with 1 ml PBS/10% FBS for either 1, 2.5, 5, 10, 20, 40, 80, or 160 min. Individual supernatants were aspirated (1 ml) and mixed with 1 ml scintillation cocktail for liquid scintillation. Cells were washed again (2 ml PBS) and then scraped from the well and transferred to a 2 ml tube, which contained 100 μl soluene 350. The mixture was homogenized, combined with scintillation cocktail up to 2 ml, and measured by liquid scintillation.
In order to wash out the [3H]-SA completely from the cell membrane, cells were incubated with various concentrations (4–20 μg/ml) of [3H]-SA for 60 min, washed (2 ml), and covered with 1 ml PBS/10% FBS for 80 min before the PBS was aspirated (first washout). Cells were covered with 1 ml PBS/10% FBS for 10 min after which the PBS was replaced with 1 ml fresh PBS/10% FBS. This process was repeated over a time period of 60 min (6× 1 ml supernatants) (second washout). All supernatants were measured by liquid scintillation as described above. Finally the cells were washed with 2 ml PBS and removed with a cell scraper and transferred to a tube for liquid scintillation as described above.
Cytotoxicity Experiments.
ECV-304 cells were seeded in 96-well plates with 2,000 cells/well in 100 μl MEM without phenol red and grown for 24 h. SA (4 μg/ml) was added, and cells were incubated for 1 h. Thereafter SA was washed out as described under “Kinetic of [3H]-SA in ECV-304 Cells.” After the second washout phase, 100 μl MEM was added, and cells were further incubated in the presence of 3 nM saporin for additional 72 h. Control cells were either incubated with a mixture of saporin (3 nM)/ saponinum album (4 μg/ml), saporin (3 nM) for the whole time or with neither saporin nor saponin.
Cytotoxicity of [3H]-SA and the toxicity-enhancing properties (10 μg/ml) on saporin (3 nM) were also scrutinized in ECV-304 cells. Control cells were either incubated with SA (4 μg/ml) and saporin (3 nM), SA (4 μg/ ml), or saporin (3 nM). After 72 h viability was determined as described above.
Statistics.
All data were analysed by Mann-Whitney U-test.
Results
Cytotoxic Experiments with [3H]-Saponinum Album ([3H]-SA).
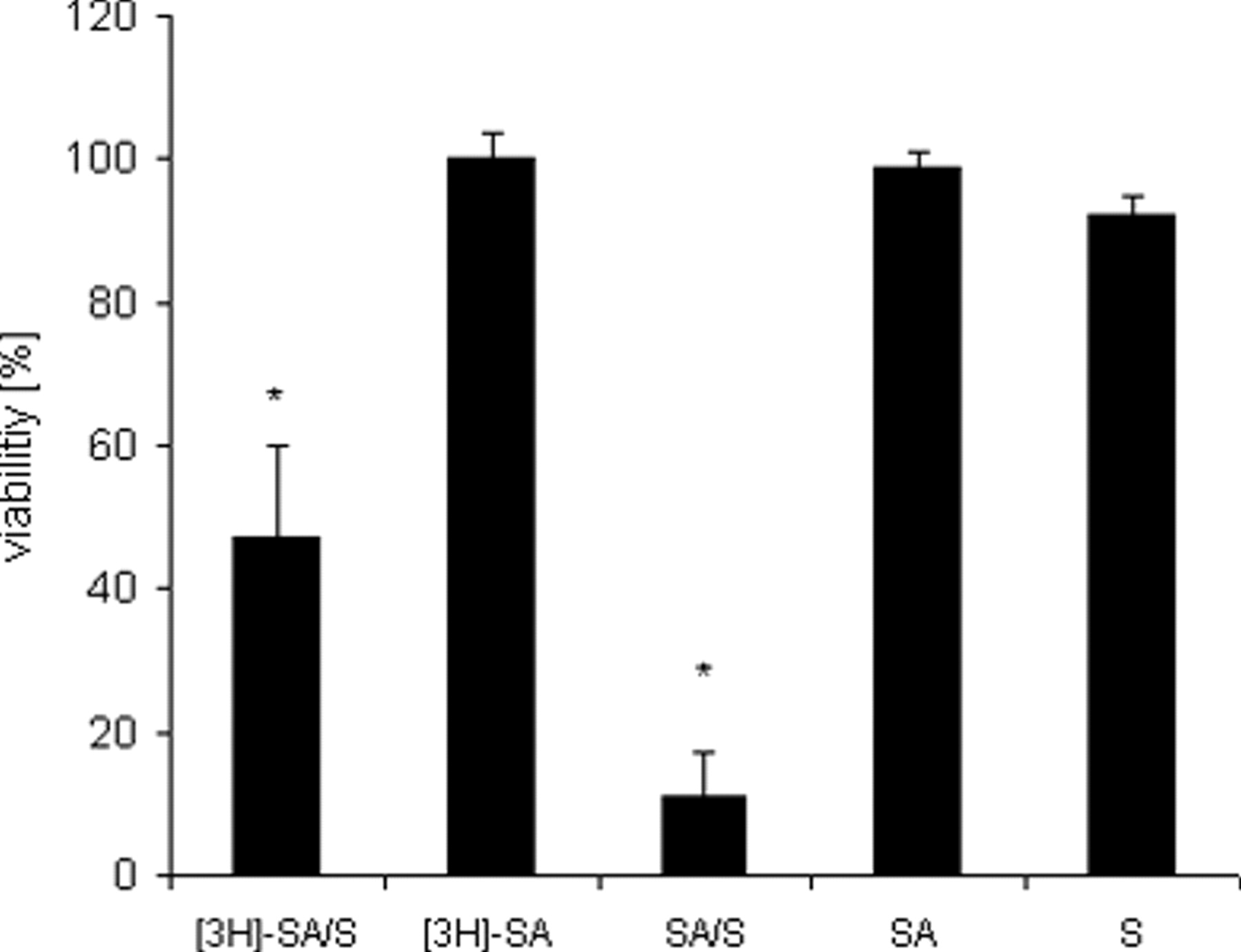
[3H]-SA (10 μg/ml) showed similar toxicity enhancing properties on saporin (S), (3 nM) as SA (4 μg/ml). [3H]-SA (10 μg/ml), SA (4 μg/ml), or saporin (3 nM) were not cytotoxic (Fig. 2).
Time Course Experiments with [3H]-Saponinum Album.
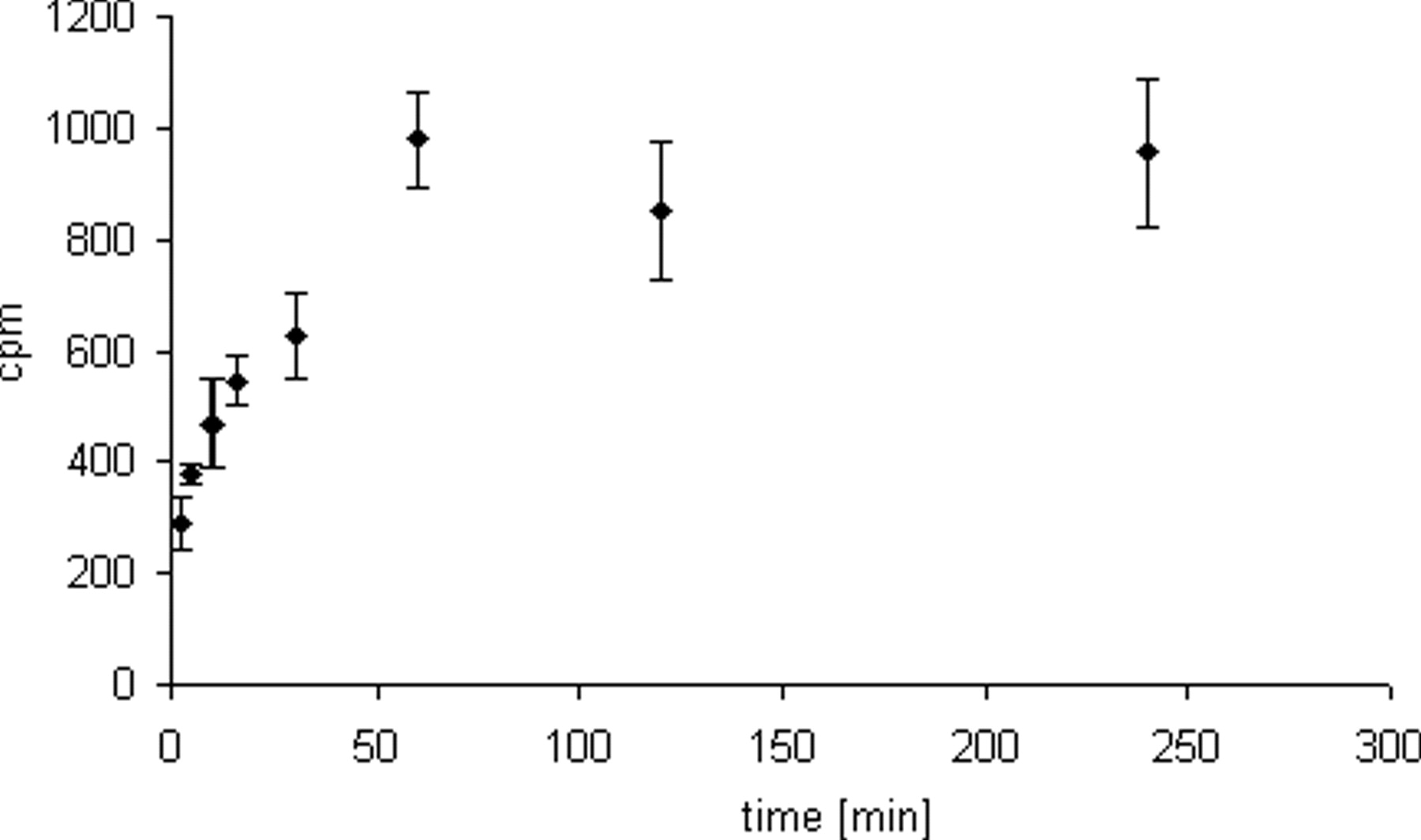
Cells were incubated with [3H]-SA (10 μg/ml) for various times ranging from 2.5 to 240 min. Treatment of cells with [3H]-SA led to an immediate insertion of saponin molecules into the cell membrane. The level of saponin molecules inserted into the cell membrane continued to increase up to 60 min. Incubation for longer time periods showed no subsequent increase in the saponin content (Fig. 3). Therefore, for further experiments it was deemed sufficient to incubate cells for only 60 min with [3H]-SA.
Kinetic of [3H]-SA in ECV-304 Cells.
We next investigated the liberation profile of [3H]-SA out of the cell. For this purpose we incubated cells with [3H]-SA (10 μg/ ml) for 60 min and then processed them as described in Materials and Methods. Following cell processing for 80 min, an equilibrium was reached between the saponin levels observed in the supernatant and cellular component (Fig. 4). Cells were then covered with PBS (10% FBS, 1 ml) for different times ranging from 1 to 160 min (first washout phase). All supernatants and the appropriate cell fraction were measured by liquid scintillation. After 80 min, an equilibrium was reached between the supernatant and the cells (Fig. 4).
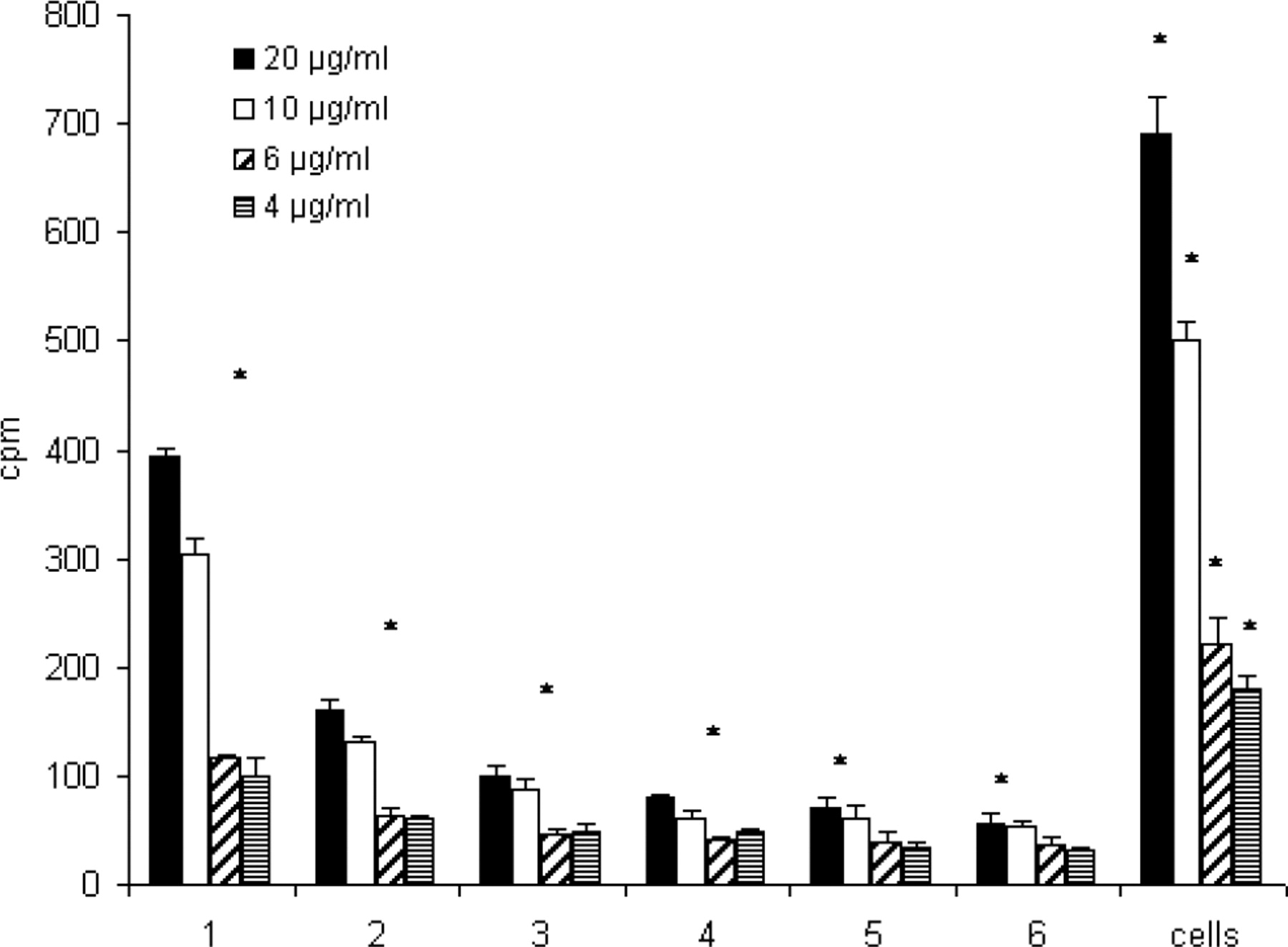
We next tried to wash out the cell associated [3H]-SA completely. For this purpose cells were incubated with various concentrations of [3H]-SA (4–20 μg/ml) for 1 h, followed by the first and second washout (see Materials and Methods).
Activity of [3H]-SA decreased in the supernatants with the number of wash steps to background levels with about 30 cpm (Fig. 5). However, it was not possible to wash out [3H]-SA completely out of the cells. A bound, durable amount of [3H]-SA was left associated with the cells following the second washout (Fig. 5, right-hand columns). For cells that were initially incubated with 20 μg/ml for 1 h, 0.43% of the total activity was associated with the cell after the second washout. For 10 μg/ml, it was 0.62%, for 6 μg/ ml 0.51%, and for 4 μg/ml 0.5%.
Cytotoxicity Experiments with SA.
ECV-304 cells were incubated with SA (4 μg/ml) for 1 h. Thereafter SA was washed out as described under “Kinetic of [3H]-SA in ECV-304 Cells.” After the second washout phase saporin (3 nM) (SA/S, left column) was added, and cells were further incubated for 72 h. The washout of SA caused no decrease of the toxicity-enhancing properties on saporin (Fig. 6). This indicates a long-lasting saponin-mediated sensitization against saporin. Control cells were either incubated with SA (72 h), saporin (S) (72 h), or SA/S (72 h) for the whole time without washing out SA.
Discussion
SA enhanced cytotoxicity of saporin 100,000-fold reflected by an IC50 of 2,000 nM without SA and 0.018 nM with SA in ECV-304 cells (4). This was also shown for a chimeric toxin composed of saporin and human epidermal growth factor (EGF) (6). The IC50 of this chimeric toxin could be reduced from 1,040 nM without SA to 0.0027 nM with SA in MCF-7 cells. This corresponds to a toxicity-enhancement factor of 385,000 (6). The SA-dependent toxicity enhancement was shown across a panel of different cell lines (5–7) indicating a common SA-dependent mechanism of toxicity enhancement of saporin and saporin-based chimeric toxins. Since toxicity of related N-glycosidases and other toxins (see Introduction) was not enhanced by SA, we assume a high specific SA interaction with the cell membrane leading to a selective toxicity enhancement of saporin and saporin-based chimeric toxins. The interaction of saponins with membranes was shown previously (1–3).
In this study we labelled SA with [3H]-acetic anhydride in order to investigate the kinetics of SA at a non-cytotoxic concentration (10 μg/ml) in a cell-culture model. [3H]-SA was shown to have cytotoxic-enhancing properties on saporin without causing any detectable self-cytotoxicity up to a concentration of 10 μg/ml (Fig. 2). This was the main prerequisite for further experiments.
We further asked if the toxicity-enhancing effect of SA upon these toxins was through a short- or long-lasting mode of activity, with specific focus on the plasma membrane. The interaction of SA with the plasma membrane and artificial membranes has been demonstrated previously (1, 2, 13, 14).
Our results indicate that there is a concentration-dependent, diffusion-controlled insertion of saponin molecules into the plasma membrane until an equilibrium (first eq.) is reached after incubation cells with [3H]-SA for 60 min (Fig. 3). The first eq. is characterized by free saponin (S) in the medium and bound saponin at the plasma membrane (M), termed the SM 1 complex.
After washing out loose bound saponin molecules from the SM 1 complex, a second equilibrium (second eq.) is reached after a further 80 min (Fig. 4) between membrane-bound saponin and the supernatant. The second eq. is characterized by a saponin-membrane complex (termed the SM 2 complex) but with lower saponin content. The saponin content of SM 2 could be further reduced by additional washing (second washout phase, see Fig. 5), resulting in the formation of a third complex termed SM 3 . SM 3 is characterized by a durable bound SA. This is possibly due to an association with cholesterol (1). We have further determined that the amount of bound saponin in the SM 3 complex was sufficient to induce a toxicity-enhancing effect on saporin (Fig. 6) as observed in control cells, which were incubated with SA/saporin from the beginning. We hypothesize that a long-lasting insertion of SA into the cell membrane caused a durable sensitization against saporin. This is highly relevant to the combined usage of saporin-based chimeric toxins and SA in tumor therapy since a prolonged susceptibility increase against these toxins could be achieved by pre-incubation with SA for 1 h.
Structure of a Gypsophila saponin from SA. Note the amphiphilic structure consisting of a hydrophobic C30 backbone (aglycone) with two hydrophilic sugar chains attached at carbon positions 3 and 28 of the aglycone. ECV-304 cells were incubated with [3H]-SA (10 μg/ml) with or without saporin (3 nM) for 72 h. Control cells were treated with SA (4 μg/ml) with or without saporin (S) (3 nM) or with only saporin (3 nM). [3H]-SA showed significant cytotoxicity-enhancing properties on saporin. All values refer to the cells that were not treated with any substances (U-test, P = 0.05). ECV-304 cells were incubated with [3H]-SA (10 μg/ml) for different times (2.5–240 min). Radioactivity in ECV-304 cells increased continuously with the time of incubation. However after 60 min a maximum was reached, indicating saturation of cells with saponin molecules. Each value represents the mean of 5 individual experimental samples. All values were significant to control cells, which were not treated with [3H]-SA (U-test, P = 0.05), n = 2. ECV-304 cells were incubated with [3H]-SA (10 μg/ml) for 60 min. Cell were covered with 1 ml PBS/10% FBS for different times (1–160 min). Activity in the supernatants (each 1 ml) and cells was measured by liquid scintillation. Note the immediate redistribution of [3H]-SA into the fresh buffer after 1 min of buffer addition. However, after 80 min the activity in the supernatant and the cells remained constant, indicating an equilibrium between membrane-bound saponin and saponin in the supernatant. Each value represents the mean of 5 individual experimental samples. All values were significant to control cells, which were not treated with [3H]-SA (U-test, P = 0.05), n = 2. ECV-304 cells were incubated with different concentrations [3H]-SA for 60 min and covered with fresh buffer for 80 min (first washout). Cells were further covered with 1 ml PBS/10% FBS for 10 min. This was repeated 6 times. Each wash fraction and cells (numbered 1 to 6) were measured by liquid scintillation. Note that the activity in the wash fractions decreased, eventually reaching background levels. Despite this, a significant amount of radioactivity could be detected within the cells after the second washout (right columns), indicating a durable insertion of saponin molecules into the cells. Each value represents the mean of five individual experimental samples. *Significant to controls, which were not treated with [3H]-SA (U-test, P = 0.05), n = 2. ECV-304 cells were incubated with SA (4 μg/ml) for 1 h, and SA was washed out as described under Material and Methods. Thereafter saporin (S) (3 nM) was added. The washout of SA caused no decrease in the toxicity-enhancing properties on saporin. Control cells were either incubated with SA (72 h), saporin (S) (72 h), or SA/S (72 h) for the whole time without washing out SA. All values refer to those cells that were not treated with any substances (U-test, P = 0.05).