
Abstract
Expression of the genes that mediate the first steps in steroidogenesis, the steroidogenic acute regulatory protein (STARD1), the cholesterol side-chain cleavage enzyme, cytochrome P450scc (CYP11A1) and 3β-hydroxysteroid dehydrogenase/Δ5-Δ4 isomerase (HSD3B), is tightly controlled by a battery of transcription factors in the adrenal cortex, the gonads and the placenta. These genes generally respond to the same hormones that stimulate steroid production through common pathways such as cAMP signaling and common actions on their promoters by proteins such as NR5A and GATA family members. However, there are distinct temporal, tissue and species-specific differences in expression between the genes that are defined by combinatorial regulation and unique promoter elements. This review will provide an overview of the hormonal and transcriptional regulation of the STARD1, CYP11A1 and specific steroidogenic HSD3B genes in the adrenal, testis, ovary and placenta and discuss the current knowledge regarding the key transcriptional factors involved.
Introduction
Steroidogenesis is a fundamental process for essential physiologic functions such as blood salt balance, carbohydrate metabolism and reproduction. Major sites for steroid hormone biosynthesis include the adrenal cortex, testis, ovary and placenta. Steroids are also generated de novo at lower levels in tissues as diverse as the kidney and the nervous system (1).
The first enzymatic step in their production is the conversion of cholesterol to pregnenolone by cytochrome P450 cholesterol side-chain cleavage (P450scc) enzyme (encoded by the CYP11A1 gene (2)) (Fig. 1). Pregnenolone is next metabolized to progesterone by select 3β-hydroxysteroid dehydrogenase/Δ5-Δ4 isomerases (3β-HSDs; encoded by HSD3B genes) (3) or in certain tissues, hydroxylated by cytochrome P450 17α-hydroxylase/17,20-lyase (P450c17α). It is from these basic steroids that all other steroid hormones are derived.
The rate-limiting step in all steroid production is the delivery of substrate cholesterol from the outer mitochondrial membrane to the inner where P450scc resides. This step is primarily mediated by the steroidogenic acute regulatory protein (StAR; encoded by the STARD1 gene) (4, 5). Steroidogenic stimuli rapidly induce the expression of StAR which then catalyzes intermembrane transfer of cholesterol to P450scc and thereby initiates steroidogenesis.
Mutations in the STARD1, CYP11A1 or HSD3B genes compromise steroid production and cause potentially lethal congenital adrenal hyperplasias (1, 6). Given the importance of these genes, it is not surprising that their expression is tightly controlled (7, 8). An understanding of the transcription factors involved is emerging thanks to important discoveries made over the last two decades. This article will briefly review the transcriptional regulation of these genes and hence, steroidogenesis in the adrenal, testis, ovary and placenta.
Regulation of Steroidogenesis
Adrenal Cortex and the Testis.
Steroid production occurs throughout life in the interstitial Leydig cells of the testes and in the zonae glomerulosa, fasciculata and reticularis of the adrenal cortex. Mature Leydig cells maintain high levels of P450scc and 3β-HSD and, in response to the pituitary gonadotropin, luteinizing hormone (LH), rapidly synthesize StAR and testosterone (9, 10).
The adult adrenal cortex also expresses high levels of P450scc, 3β-HSD and StAR (11). Glomerulosa cells generate steroid with stimulation by angiotensin II (AII) and potassium, while cells in the other cortical layers are under the control of adrenocorticotropin (ACTH). While the key hormone receptors change between layers, the transition is not abrupt—the zona glomerulosa does possess receptors for ACTH while the fasciculata exhibits sensitivity to AII, though not potassium.
The steroids produced also change between zones. Glomerulosa cells generate the mineralocorticoid aldosterone in response to AII and potassium. Cell types in the fasciculata and reticularis primarily synthesize glucocorticoids, except in the reticularis of the human and select primates where a lack of 3β-HSD results in the synthesis of dehydroepiandrosterone (DHEA) and DHEA sulfate. Another species-dependent difference is that corticosterone is the primary glucocorticoid product of the rodent adrenal, not cortisol, due to an absence of significant levels of P450c17α.
One similarity between the testis and the adrenal is that both LH and ACTH stimulate steroidogenesis chiefly through the cAMP second messenger pathway. AII stimulation on the other hand follows a protein kinase C (PKC)-dependent route, mimicked by phorbol ester, and as with potassium, involves Ca2+-calmodulin-dependent kinase as well (12).
Ovary.
In the ovary steroidogenesis takes place primarily in the somatic granulosa and theca cells surrounding the oocyte of the developing follicle (13). The theca cells reside in the outermost layer of the follicle and generate androgens de novo. In most mammals these androgens are mainly aromatized to estradiol after diffusing across the basement membrane to the granulosa layer, but can be aromatized in the theca of some species. LH stimulates androgen production whereas a second gonadotropin, follicle stimulating hormone (FSH), stimulates the expression of aromatase in the granulosa and thereby estrogen synthesis. Interstitial glands found in some species also bear the capacity to produce steroids (14).
Later in folliculogenesis, the ovulatory surge in LH leads to a drop in estradiol production (transient in some species) and a dramatic increase in progesterone production by the residual luteinizing granulosa and theca cells (13). This new function of the cells accompanies terminal differentiation or luteinization and, for granulosa cells, relies upon an increase in LH receptors just prior to the surge. Then with the dramatic rise in LH, granulosa cells acquire the ability to make progesterone from cholesterol, coincident with the appearance of StAR (15).
The major steroid product of the newly formed corpus luteum is progesterone which supports the uterine endometrium, a function essential for implantation of the embryo and maintenance of pregnancy. Resident luteal cells express high levels of STARD1, CYP11A1 and HSD3B type 1 (rodents and most species) or type 2 (humans) in order to turn out high levels of the steroid (15). The corpus luteum is maintained by trophic stimuli, like chorionic gonadotropin in human (hCG) and prolactin in rodents, if implantation occurs. Otherwise it regresses allowing for another reproductive cycle. Luteal regression or luteolysis can be stimulated in different species by PGF2α or by the preovulatory prolactin surge in rodents (16).
Placenta.
The timing, cellular distribution and importance of de novo steroid synthesis by the placenta vary greatly between species. In humans, cytotrophoblast-derived syncytiotrophoblasts in the placenta take over the responsibility for progesterone and estrogen production from the corpus luteum near the end of the first trimester of pregnancy. Since the human placenta lacks P450c17α, estrogens are not synthesized de novo but rather originate from circulating maternal and fetal androgens (17). Syncytiotrophoblasts express mRNAs and protein derived from the genes for CYP11A1 and a placental HSD3B isoform, but uniquely in the human, not STARD1—some suggest that its homologue, MLN64 assumes its function (18, 19). Amazingly, progesterone synthesis in the human reaches 250 mg/day by the end of pregnancy (circulating levels of 130 ng/ml), illustrating the tremendous steroidogenic capacity of these cells in aggregate (20). While severe deficits in placental P450scc activity do not cause an early loss of pregnancy (1), there are no known diseases involving the loss of placental HSD3B, suggesting that its loss is lethal in utero.
In contrast to the human, there is no luteoplacental shift in mice and rats during pregnancy. Instead, rodents require luteal output of progesterone and estrogen through to parturition (21). Still, the decidua and placenta are sites of progesterone synthesis. In mice de novo steroid production occurs in the maternal decidua capsularis around the time of implantation. Later, the giant trophoblast cells of rats and mice constitutively produce progesterone. These cell types all express STARD1, CYP11A1 and a placental 3β-HSD isoform (21–24). The importance of de novo placental steroid production at least in the mouse is unclear since knockouts of STARD1 and CYP11A1 survive to term (1).
Hormonal Control of STARD1 Gene Expression
Numerous extracellular factors have been shown to alter StAR protein and steroid synthesis. Below we summarize the key molecules that alter steroidogenesis, primarily focusing on those for which evidence exists of their regulating StAR production at the level of transcription.
Regulation of STARD1 mRNA in the Adrenal Cortex.
The production of StAR correlates with steroidogenic status in the adrenal cortex. Steroidogenic stimuli such as AII, ACTH and the cAMP analogue dibutyryl cyclic AMP [(Bu)2cAMP] all increase STARD1 mRNA levels.
The effect of ACTH on STARD1 mRNA has been shown in model systems such as the rat adrenal and cultured human and bovine adrenocortical cells, which tend to approximate fasciculata-like cells (25–28). AII stimulates STARD1 mRNA in bovine adrenocortical cells and human H295R adrenocortical carcinoma cells but potassium has no effect (28, 29). However, potassium can induce StAR protein and steroid synthesis in H295R and bovine glomerulosa cells, possibly involving post-transcriptional regulation and phosphorylation of StAR (30–33). Phorbol ester also can affect StAR activity in these cell types, but not STARD1 mRNA, suggesting that AII acts through crosstalk with the cAMP pathway to stimulate STARD1 gene expression.
Calcium a major signaling molecule in the adrenal increases while atrial natriuretic peptide (ANP) (34) opposes STARD1 transcription and aldosterone production in bovine glomerulosa cells (29, 35). Insulin-like growth factor I (IGF-I), which can be locally produced by adrenal cells or come from serum (36), by itself stimulates STARD1 mRNA levels and enhances the effect of ACTH on STARD1 in bovine adrenal cells (28). This is consistent with the known ability of IGF-I to enhance cAMP-mediated steroidogenesis in the adrenal and gonads (37–39). There are conflicting reports on how STARD1 expression in H295R cells is affected by activin A, which apparently promotes a shift in steroid production from androgens and glucocorticoids to aldosterone (40, 41). The cytokine bone morphogenetic protein (BMP)-6 also increases STARD1 mRNA and promoter activity in H295R cells (41). Other cytokines, transforming growth factor β1 (TGFβ1) and interleukin-10 (IL-10), and the adipokine leptin inhibit while leukemia inhibitory factor (LIF) increases STARD1 mRNA accumulation in adrenocortical cells in keeping with their modifications of adrenal steroidogenesis (28, 42–45). Cholesterol sulfate, known to interfere with the conversion of cholesterol to pregnenolone (46), reduces basal and cAMP-stimulated STARD1 mRNA and promoter activity in H295R cells (47) whereas lipoproteins stimulate STARD1 in mouse Y1 adrenocortical tumor cells (48, 49).
Regulation of STARD1 mRNA in the Testis.
LH stimulates STARD1 mRNA and protein production in Leydig cells and its effect is mimicked by its homologue hCG and cAMP analogues (50–52). Numerous other hormones and locally produced or serum-borne growth factors also induce STARD1 mRNA, including epidermal growth factor (EGF) and TGFβ through the EGF receptor and in certain culture conditions, and IGF-I by themselves or through augmentation of the hCG response (52–55). Other positive regulators of steroidogenesis and STARD1 include progesterone under certain conditions (56), short-term thyroid hormone exposure (57, 58), fibroblast growth factor (54), colony-stimulating factor-1 (54), activin A (59), 9-cis-retinoic acid (60) and Mullerian inhibiting substance (61). Levels of STARD1 mRNA also rise in immature rat Leydig cells in response to TGFβ (54, 62) and IL-1α (54, 63), but have only limited effects on mature cells. Corticotrophin releasing hormone (CRH) stimulates the protein in mouse MA-10 Leydig tumor cells (64). Repressors of STARD1 mRNA in Leydig cells include the synthetic glucocorticoid dexamethasone (65), leptin (66) and cytokines tumor necrosis factor α (TNFα) (67, 68), LIF (69) and interferon γ (70). Gene expression is also indirectly affected by prolactin in a biphasic manner. Low prolactin levels increase LH receptor (LHR) gene expression and consequently hCG-stimulation of STARD1 and steroid production, whereas much higher concentrations reduce LHR mRNA and the effects of hCG (71). In summary, numerous serum-derived and locally produced growth factors, cytokines, and steroids regulate STARD1 expression in a pattern paralleling their effects on the production of steroids, especially progesterone.
Interestingly, StAR is also found in luminal rat and human Sertoli cells, albeit at a much lower level (72, 73). Its expression is increased by cAMP and FSH (72, 74) and inhibited by IL-1β (75). Since there is no detectable P450scc basally, the role of StAR in this cell type is unclear.
Regulation of STARD1 mRNA in the Ovary.
LH, hCG and other cAMP signals stimulate STARD1 mRNA expression in theca, luteinized granulosa and luteal cells (76–82). During follicle development in the adult ovary, STARD1 is expressed in the theca cells under the influence of LH, but not in the granulosa cells of most species until the periovulatory LH surge (76, 77, 83, 84). With the LH surge, STARD1 is dramatically upregulated, particularly in the granulosa layer, and continues to be strongly expressed in the luteinized theca and granulosa cells that comprise the corpus luteum (73, 76, 77, 85, 86). STARD1 expression also rises in steroidogenic granulosa cells in the cumulus layer surrounding the ovulated oocyte (87).
FSH also induces STARD1 mRNA in granulosa cells in culture, but there is no evidence to support FSH action in the animal (76, 77, 88, 89). The appearance of StAR in the granulosa cell in vivo appears to be due to the actions of LH. Still, FSH is a good tool to investigate the actions of cAMP in this cell type in culture.
Various growth factors, many of which are produced locally, also directly induce or modify FSH and LH stimulation of STARD1 gene transcription in a species-specific manner consistent with their observed changes in steroid production. One of the best-studied factors is IGF-I, which acts as a biological amplifier of several gonadotropin-stimulated endpoints including steroidogenesis (90). IGF-I modestly drives STARD1 mRNA production in cultured granulosa cells as well as enhancing the effect of gonadotropin and cAMP (80, 88, 91). Other positive regulators of STARD1 mRNA include hCG acting via LH receptors (77), IGF-II (92), insulin (80, 88, 92) and low doses of leptin (93). StAR protein is also increased by estradiol in rabbit granulosa, but its effect on STARD1 mRNA is unknown (94). STARD1 mRNA is further induced by amphiregulin in cumulus granulosa cell-oocyte complexes (95) and LH (78, 79), stem cell factor (SCF) with IGF-I (96), and insulin (79) in cultured theca cells.
Numerous peptides suppress basal or gonadotropin effects on ovarian steroidogenesis and STARD1 expression. These factors can be secreted by the oocyte, granulosa or theca cell to provide paracrine/autocrine signals and may suppress the premature expression of STARD1 in granulosa. Unlike Leydig cells, EGF suppresses FSH- or cAMP-stimulated STARD1 gene expression in granulosa (91, 97). Other repressors include TGFβ in theca cells (98), high doses of leptin (93) in granulosa cells, and cholesterol sulfate in a granulosa tumor cell line (99). TGF-β negatively affects STARD1 mRNA in cultured bovine granulosa cells (100), but enhances FSH-stimulated StAR protein in rat granulosa cells (101). In granulosa cells, several BMPs are negative regulators of FSH-stimulated mRNA or protein levels including BMP-2 (102), −4 (102, 103), −5 (104), −6 (102, 105, 106), −7 (102, 107) and −15 (108). Since BMP-4, −6 and −15 can reduce cAMP production or FSH receptor mRNA levels, their negative effects on STARD1 mRNA may in part be secondary to impaired FSH signaling (103, 105, 106, 108). BMP-4, −6 and −7 also decrease basal mRNA in theca cells (109).
Regulation by follicular product growth differentiation factor (GDF)-9 is species-dependent, increasing basal STARD1 levels in cultured granulosa cells from the mouse but not the human (110, 111). GDF-9 further inhibits cAMP-stimulated mRNA and protein accumulation and promoter activity in human luteinized granulosa cells (111). This protein also opposes basal steroidogenesis and StAR protein levels in human theca cells (111).
In luteal cells, STARD1 content is stimulated by luteotroph PGE2 (112). Factors that induce functional regression of the corpus luteum like PGF2α (81, 113, 114) and TNF-α (115) decrease STARD1 expression in this cell type.
Regulation of STARD1 mRNA in the Rodent Placenta.
Midgestational giant trophoblast cells are the primary site of STARD1 expression in the rodent placenta. The hormones regulating placental STARD1 expression in vivo are currently unknown, but treatment of murine giant trophoblast cells with a cAMP analogue increases StAR protein levels and stimulates STARD1 promoter activity (116). Interestingly though, progesterone production by mouse and rat giant trophoblast cells in culture is independent of cAMP stimuli (117, 118), suggesting that steroid output is limited by existing, cAMP-insensitive P450scc levels instead of cholesterol supply to the enzyme.
Transcriptional Control of the STARD1 Gene
Studies aimed at understanding the complex regulation of the STARD1 gene have focused on the highly conserved proximal region of the promoter (Fig. 2). Two areas within this region in particular, encompassing base pairs −119 to −95 and −81 to −58 in the human, largely govern stimulation of STARD1 expression. Here we consider the cis- and trans-acting factors that bind to these regions as well as others to regulate this key gene in steroidogenesis.
Steroidogenic Factor 1 (SF-1) and Liver Receptor Homologue 1 (LRH-1) Family.
The orphan nuclear receptors SF-1 (NR5A1) and LRH-1 (NR5A2) are present in all steroidogenic cells in the gonads and adrenal (except for ovarian theca and steroid-producing interstitial cells in the case of LRH-1). The two proteins bind the same DNA sequence (119) and regulate critical genes in the reproductive axis and steroidogenesis (120), including STARD1. Multiple conserved NR5A recognition sequences have been identified in the human STARD1 promoter with at least two functional elements at −105 to −95 and −42 to −35 and one at −926 to −918, relative to the transcriptional start site (121). One or more of these sites is needed for full basal and/or cAMP responsiveness in Leydig and luteinized human granulosa cells in culture (116, 121–124). Mutation of the equivalent −105 to −95 site or a more distal element at −137 to −132 but not the most proximal element in the murine promoter dramatically decreases the basal and EGF-stimulated response, but apparently not the fold increase elicited by EGF in mLTC-1 mouse Leydig tumor cells (55). The −105 to −95 SF-1 site may also be critical for AII and (Bu)2cAMP stimulation of the human STARD1 promoter in H295R cells (29).
The individual roles of NR5A proteins are still being determined as early studies carried out prior to the discovery of LRH-1 in steroidogenic tissues focused on SF-1 (125, 126). Selective knockout of SF-1 causes a catastrophic loss of StAR expression in the testes of transgenic mice (127). In mLTC-1 cells, SF-1 increases basal activity and the fold response of the promoter to (Bu)2cAMP with or without IGF-1 (53). Chromatin immunoprecipitation (ChIP) assays further confirm increased SF-1 association with the mouse STARD1 promoter in response to cAMP stimulus in MA-10 Leydig tumor and murine granulosa cells (128).
In mouse Y1 adrenocortical cells and clonal cell types (nonsteroidogenic cells), basal and cAMP-stimulated rat and human promoter activities also involve SF-1 (125, 129, 130). Accordingly, ACTH recruits SF-1 to the promoter in primary murine adrenal cells (131). Recent RNAi experiments in H295R cells confirm a role for SF-1 in determining the magnitude of STARD1 levels, but disputes its importance in cAMP-induction (132). On the other hand, impaired SF-1 activity eliminates 8-bromo-cAMP induction of steroid synthesis and knocks down basal STARD1 mRNA levels by 75% in a mutant Y1 cell line (133).
Expression of the other NR5A family member LRH-1 in granulosa cells is required for ovulation (134). Like SF-1, it binds the −105 to −95 site and transactivates the STARD1 promoter in clonal and human granulosa tumor cells (135, 136). However, the proximal NR5A sites do not participate in FSH-stimulated STARD1 promoter activity in granulosa cells (89, 116, 137). This changes with luteinization. Upon hCG treatment, LRH-1 associates with the endogenous STARD1 promoter in ovarian tissue in mice (134). Mutagenesis of the −105 to −95 SF-1 element now dramatically reduces promoter activity (116). Targeted loss of LRH-1 in granulosa cells also disrupts STARD1 mRNA levels and progesterone production in hCG-treated transgenic mice (134). Since with luteinization LRH-1 levels exceed those of SF-1 (138–140), LH-insensitive regulation of the STARD1 gene in luteal cells is likely managed in part through LRH-1, but this is not a settled point (116).
While SF-1 and LRH-1 have important functions in regulating STARD1 gene expression, their precise roles and sites of action may require further revision given recent findings regarding a second orphan nuclear receptor family, NR4A.
NR4A Family.
NR4A transcription factors represented by Nur77 (NGFI-B; NR4A1), nur related protein-1 (Nurr1; NR4A2) and neuron-derived orphan receptor 1 (Nor1; NR4A3) bind sequences similar to SF-1/LRH-1 sites, called NGFI-B response elements (NBREs) (141). NR4A proteins have been described in cAMP and phorbol ester-stimulated human granulosa tumor cells, human corpora lutea and rat granulosa cells in post-surge preovulatory follicles (142–144). In H295R adrenocortical cells, AII and potassium rapidly induce Nur77 and Nurr1 (145). ACTH and (Bu)2cAMP induce Nur77 production in all layers of the murine adrenal cortex, Y1 cells and primary human adrenal cell cultures (146–148). LH also upregulates Nur77 in primary rat Leydig and MA-10 and K28 mouse Leydig tumor cells (149, 150).
Nur77 is the predominant NR4A protein in steroidogenic cells and its expression activates the murine STARD1 promoter to various degrees in Y1 and Leydig tumor cells (67, 149, 151). It recognizes the proximal NRBE, which coincides with the −105 to −95 SF-1 site, and cAMP-stimulation increases its association with the region of the STARD1 promoter containing this element in Leydig cells (149). This implies that the effects on cAMP and EGF stimulation observed with mutation of this site may more accurately reflect the actions of Nur77 not SF-1 (55, 149). For instance, promoter stimulation by Nur77 is decreased by mutation of the site in MA-10 cells (152). Consistent with the aforementioned studies of the −95 site in granulosa cells, Nur77 also has little effect on basal promoter activity in human granulosa tumor cells (142).
However, the reported activities of SF-1/LRH-1 and Nur77 may not be duplicative. For instance, dexamethasone repression of cAMP stimulation of the murine STARD1 promoter targets the −95 site in MA-10 cells and causes reduced association of the proximal promoter with Nur77, not SF-1 (153). Conversely, Nur77 may indirectly inhibit SF-1 binding (154). Thus, Nur77 may supplant NR5A protein binding to this site to facilitate cAMP stimulation of the STARD1 gene in Leydig cells (152). SF-1 participation in stimulated activity may involve a different site of action.
DAX-1.
Another orphan nuclear receptor, DAX-1 (dosage-sensitive sex reversal, adrenal hypoplasia congenita, critical region on the X chromosome, gene 1; NR0B1), represses the STARD1 gene and its expression pattern in the body mirrors that of SF-1 (155). Steroidogenic stimuli downregulate DAX-1, coincident with increases in STARD1 gene expression in primary glomerulosa and MA-10 cells (151, 156). Luteolytic PGF2α and dexamethasone increase DAX-1 levels while decreasing STARD1 mRNA in rat ovaries and primary adrenocortical cells, respectively (131, 157). Constitutive production of StAR and steroid in Y1 and rat R2C Leydig tumor cells is linked to low levels of DAX-1 (158, 159). Exogenous DAX-1 inhibits StAR in R2C cells and basal and forskolin-driven STARD1 promoter activity and endogenous gene expression in Y1 cells (157–160). This transcription factor also suppresses Nur77 and SF-1 activation of the STARD1 promoter in Leydig cell lines (151, 161), as well as LRH-1 activation of the promoter in human granulosa tumor cells (136). Naturally occurring mutations in DAX-1 disrupt this inhibition of the promoter in Y1 cells (162). As well, RNAi knockdown of DAX-1 in MA-10 cells increases basal and (Bu)2cAMP and phorbol ester-stimulated StAR production (151).
The mechanism of DAX-1 repression is unique. While DAX-1 lacks a consensus DNA-binding domain, it is proposed to bind specific DNA hairpin structures. Such hairpins are predicted to form between −61 to −27 in the human (160) and −44 to −20 in the murine promoter (151). Both ChIP and in vitro DNA binding assays suggest that binding of this hairpin by DAX-1 inhibits murine STARD1 expression in MA-10 cells and that the loss of binding with (Bu)2cAMP or phorbol 12-myristate 13-acetate (PMA) derepresses promoter activity (151). The putative formation of an additional DAX-1 complex due to a −33 C/T polymorphism in the human promoter reduces forskolin and AII-stimulated promoter activity in Y1 and H295R cells, respectively (163).
DAX-1 can also repress Nur77, SF-1 and LRH-1 transactivation through direct protein-protein interactions (164, 165). Thus, DAX-1 inhibits STARD1 gene activity at multiple levels whereas stimulation of the promoter partly derives from declines in DAX-1 levels.
GATA Family.
The role of GATA factors is very significant. The GATA site located at −63 to −58 in the human and −66 to −61 in the mouse is highly conserved. This region efficiently binds recombinant GATA4 and GATA6 (137). The site comprises about 20% of the cAMP responsiveness of the murine promoter in MA-10 Leydig cells (123) and 30–35% of the maximal FSH response in cultured granulosa cells (89, 137). Mutation of the murine sequence also reduces basal and EGF-stimulated activity in mLTC-1 cells (55). Correspondingly, steroidogenic tissues express select GATA factors, such as GATA6 in the adult adrenal cortex (166). Overexpression of this factor drives human promoter activity in adrenocortical cells (167). Results in a nonsteroidogenic cell line indicate that stimulation of the gene by GATA6 is sensitive to DAX-1 and depends on synergy with SF-1 (167).
Somatic cells in the ovary and testis contain both GATA4 and GATA6 (168). GATA4 overexpression drives the murine STARD1 promoter in Leydig tumor cells and is enhanced by co-transfection with the homeobox transcription factor distal-less homeobox 5 (Dlx5) (169). GATA4 also augments stimulation of the promoter by (Bu)2cAMP or (Bu)2cAMP with IGF-1 in mLTC-1 cells (53). Accordingly, association of GATA4 with the endogenous proximal promoter in MA-10 cells increases within 15 minutes of treatment with 8-bromo-cAMP (128).
While both overexpressed GATA4 and GATA6 effectively drive porcine STARD1 promoter activity in granulosa cells (170), which GATA protein actually directs, STARD1 expression may vary with the level of differentiation. Association of GATA4 with the endogenous proximal promoter immediately increases in murine granulosa cells following an ovulatory hCG stimulus coincident with the dramatic induction of STARD1 in this cell type (128). With luteinization, however, GATA4 levels decline in vivo (170–173), leaving GATA6 as the likely family member that facilitates luteal STARD1 gene transcription. A recent RNAi study in luteinizing porcine granulosa cells lends insight into its function (174). The authors found that reduction of GATA4 levels enhances cAMP-stimulated STARD1 mRNA accumulation, but not when GATA6 is also reduced. Thus GATA6 may be a more potent inducer of STARD1 expression, paralleling the observed increases in StAR and steroid output with differentiation.
CCAAT/Enhancer Binding Protein β (C/EBPβ).
C/EBP family members regulate both proliferation and differentiation depending upon their transcriptional partners (175) and are present in adrenal and gonadal tissues (176–178). One member, C/EBPβ, is upregulated in the ovary by gonadotropins, cAMP and luteinization (89, 176, 177, 179) and is required for ovulation (180).
C/EBP recognition sequences localize near the GATA and SF-1 sites in the proximal STARD1 promoter. One highly conserved site, denoted C1 in the mouse, is located at −119 to −110 in the human. Other sites are more proximal and their location, number and sequence vary by species. A more proximal C/EBP site exists in humans at −50 to −41 (179). A slightly different proximal site conserved in ruminants and pigs resides at −58 to −49 in the porcine promoter and is bound by C/EBPβ in porcine granulosa cell nuclear extracts (137).
The mouse STARD1 promoter has two other C/EBP sites downstream of C1, dubbed C2 and C3. In transfected nonsteroidogenic COS-1 epithelial cells, both C1 and C2 are required for SF-1 transactivation of the murine promoter (181). Moreover, recombinant C/EBPβ and SF-1 physically interact with each other in the presence of a DNA fragment of the proximal murine STARD1 promoter (181). However, mutation of either C1 or C2 only reduces the magnitude of basal and (Bu)2cAMP-stimulated promoter activity in MA-10 cells, suggesting that the cAMP response is not driven from either site (181). Similar observations were made for C1 in regard to EGF stimulation in mLTC-1 cells (55). As might then be predicted, overexpression of C/EBPβ only affects the size of the response to (Bu)2cAMP and (Bu)2cAMP with IGF-1, not the relative increase in reporter gene products (53). Moreover, cAMP-stimulation of MA-10 cells does not increase the level of C/EBPβ found at the endogenous STARD1 promoter (128).
In contrast to Leydig cells, gonadotropin does recruit C/EBPβ to the proximal region of the murine promoter in granulosa cells in vivo (128). An early study of the murine promoter implicated C/EBP binding to the C3 site as an important contributor to FSH-induction of the STARD1 promoter in rat granulosa cells (89). However, a more recent paper by the same authors suggests that although C/EBP binds the C3 site with FSH stimulation, selective nucleotide mutagenesis supports cAMP response element binding protein (CREB) rather than C/EBP as the participating factor (see CREB section below) (116). With luteinization, cAMP-independent murine promoter activity in rat granulosa-luteal cells now becomes partly dependent on the C1 C/EBP site (116).
Mutation of both C/EBP sites in the human (−119 to −110 and −50 to −41) affects the magnitude of basal and cAMP-stimulated STARD1 promoter activity in human luteinized granulosa cells with little change in the fold-response to cAMP (179). In overexpression studies, C/EBPβ cooperates with GATA4 to stimulate STARD1 promoter activity in porcine granulosa and clonal cell types (137, 173), and a physical interaction is suggested in MA-10 cells (182). In porcine granulosa cells, maximum induction of the porcine promoter by FSH with or without IGF-I depends upon the most proximal −58 to −49 C/EBP site in conjunction with the adjacent GATA site (137). Thus, C/EBPβ may serve to support basal and stimulated gene expression.
CREB Family.
CREB family proteins such as CREB, CRE modulator (CREM) and activating transcription factors (ATFs) typically mediate cAMP responses via cAMP response elements (CREs) following phosphorylation (183). Although the STARD1 promoter is highly sensitive to cAMP stimuli, it lacks a consensus CRE (184). However, CRE half-sites are present in most species. The −110 to −67 region of the murine STARD1 promoter contains three, designated CRE1, CRE2 and CRE3. Overexpression of CREB in MA-10 and Y1 cells increases basal and the size and fold activation of the (Bu)2cAMP response of the murine STARD1 promoter activity, whereas overexpression of inhibitors of CREB activity, nonphosphorylatable CREB or a dominant negative, lowers these activities (124, 185). Whereas all three CRE sites may contribute to cAMP-responsiveness of the promoter in MA-10 cells, the −81 to − 78 CRE2 site is clearly the more important (124, 185). While CREMτ can mimic the action of CREB, current evidence indicates that CREB is the primary family member that binds the CRE2 site, perhaps as a dimer with ATF-1 (185, 186). On the other hand, CREM may play a vital role in adrenal activation of the STARD1 promoter, as depletion of CREM but not CREB by RNAi blocks cAMP-induced STARD1 expression in H295R cells (132).
Various degrees of inhibition of basal and (Bu)2cAMP-induced activity are observed with a CRE1 mutation that includes the alteration of a base shared with the overlapping SF-1 site or when SF-1 and CRE sites are otherwise mutated in combination (185). Perhaps unsurprisingly then, co-expression of CREB and SF-1 additively increases the level of activity in cAMP-stimulated MA-10 cells (124).
One complication in studies on the role of the CRE2 site is that it falls within the −81 to −72 C3 C/EBP site (89) and has a one base-overlap with C2. Cellular specificity of the murine proximal promoter was clarified by landmark work that employed ChIP assays with the murine STARD1 promoter in both cAMP-stimulated MA-10 Leydig tumor and mouse periovulatory granulosa cells collected following hCG stimulation in vivo (128). In response to cAMP stimulus, ChIP assays revealed CREB/CREM but not C/EBPβ increased association with the proximal STARD1 promoter in MA-10 cells within 30 minutes (128). A second study using ChIP indicated that upon stimulation, phosphorylated CREB levels strongly rise at the proximal promoter, whereas total CREB levels remain constant (186). The discrepancy between the two studies as to total CREB association with the promoter may be explained by the use of a more sensitive detection tool, real-time PCR, in the Hiroi study (186). On the other hand, Hiroi and colleagues observed an increase in levels of C/EBPβ at the proximal promoter in granulosa cells within 1 hour of hCG, with a more delayed association of CREB (128), implying a primacy of C/EBPβ in this case.
However, as intimated before, a selective mutation of murine C3 that does not alter the CRE2 site nonsignificantly depresses FSH-responsiveness in granulosa cells, whereas a mutation that disrupts the CRE2 site as well as a CREB inhibitor has pronounced effects on stimulated promoter activity (116). Mutational analysis also indicates that GATA4 and CREB cooperate through this site (89, 116). With luteinization, CREB inhibition now has no effect on the constitutive promoter activity that typifies STARD1 expression in rodent granulosaluteal cells (116). Thus, changes in CREB binding to the CRE2/C3 element contribute to cell- and sex-specific regulation of the STARD1 gene and are important for full responsiveness to FSH in rodent granulosa cells (116).
Activator Protein 1 (AP-1) Family and Their Role in STARD1 Gene Regulation in Leydig Cells.
The AP-1 family of immediate-early response genes includes c-Jun, JunB, JunD and Fos family members c-Fos, FosB, Fos-related antigen (Fra) 1 and Fra-2 and affects a diversity of cellular processes including STARD1 expression (187, 188). PMA increases c-Jun in cAMP-stimulated MA-10 cells (189). c-Fos and Jun family members also rise in response to PMA, hCG and to a lesser extent, EGF in Leydig cells (190). The question of which AP-1 proteins are induced by cAMP in Leydig cells has recently been revisited (152, 189, 191).
Extensive research has focused on the role of AP-1 proteins on the murine promoter in Leydig cell models. Overexpression of FosB and Jun proteins enhances while Fra-1 and Fra-2 decrease basal STARD1 promoter activity in MA-10 cells (188, 191). As well, c-Jun stimulates basal promoter activity above that achieved by CREB and Nur77 (152, 191). Basal is also elevated by c-Jun and JunD while Fra-1 is slightly inhibitory in mLTC-1 cells (55). Co-expression of both c-Jun and Fra-2 attenuates the level of promoter activity induced by c-Jun alone in MA-10 cells (188). Transfectants expressing c-Fos register a modest enhancement or inhibition of basal promoter activity in both mLTC-1 and MA-10 cells (55, 152, 188, 191). However, increasing concentrations of c-Fos dose-dependently increase promoter activity in MA-10 cells (188). Decreased expression of c-Jun, c-Fos and even Fra-1, either by antisense targeting or expression of dominant negative forms of these factors, reduce basal promoter activity (53, 188). In the case of c-Jun, activity is ablated.
AP-1 proteins also have important roles in PMA and cAMP-stimulated promoter activity. The mouse promoter contains a fairly well-conserved AP-1 or TPA response element (TRE) at −81 to −75 overlapping the CRE2 site (188, 191) that binds factors such as c-Fos, Fra-2 and JunD in nuclear extracts from (Bu)2cAMP-stimulated MA-10 cells (188). Analyses by ChIP show that while the absolute level of endogenous c-Jun associated with the proximal promoter is unchanged with (Bu)2cAMP-stimulation in MA-10 cells (186), the level of phosphorylated c-Jun acutely increases (191). Expression of FosB or Jun family members increases the magnitude of the cAMP response of the murine promoter in transfected MA-10 cells, but reduces the fold increase relative to basal (53, 188, 191). The stimulatory effect of (Bu)2cAMP alone or when combined with IGF-I is attenuated in the presence of a dominant negative form of c-Jun (53). c-Jun also additively increases stimulation by NR4A proteins from the −95 SF-1/NBRE element and synergizes with SF-1 and LRH-1 from the proximal −45 SF-1 element in MA-10 cells (152). However, c-Jun and JunD have little or no effect on EGF stimulation (55).
Phorbol ester stimulation of STARD1 expression also coincides with the recruitment of phosphorylated c-Jun to the proximal promoter in MA-10 cells (189). Again, the magnitude but not the fold response to PMA rises with c-Jun expression and targeted inhibition or loss of c-Jun blocks this response (189). On the other hand, while c-Fos and Fra-1 attenuate the response to EGF (55) and c-Fos, Fra1 and Fra2 inhibit cAMP-stimulated promoter activity in Leydig tumor cells (188, 191). At the same time, cAMP acutely increases levels of c-Fos and phosphorylated c-Fos as well as the association of the phosphorylated protein with the proximal promoter (191).
So both AP-1 and CREB factors have roles in the cAMP response, but knowledge of what they are is complicated by the fact that the murine AP-1 and CRE2 sites completely overlap. Mutation of the CRE2/AP-1 site generally decreases basal and stimulated promoter activity in Y1 and Leydig cell lines (55, 114, 123, 181, 186, 188). As revealed by ChIP assays, the time courses of binding to this promoter region by AP-1 and CREB differ with cAMP stimulation in MA-10 cells (191). Upon stimulation phosphorylated c-Jun, c-Fos, and CREB initially increase association with the region encompassing the CRE2/AP-1 site, then phosphorylated c-Jun and c-Fos association decline whereas that of CREB remains high. These results suggest that following cAMP stimulation, occupancy of the CRE2/AP-1 region in MA-10 cells favors phosphorylated CREB with time (191).
Mutation of the CRE2/AP-1 site does not abolish all c-Jun-mediated activity or its ability to cooperate with Nur77 (152). Thus, c-Jun may further act through another promoter site or different mechanism, such as a direct interaction with Nur77 (152).
Functionally, c-Fos represses CREB stimulation of the STARD1 promoter in MA-10 cells in the presence or absence of a cAMP analogue (191). Co-expression of CREB and c-Jun also attenuates the increases in basal and cAMP-stimulated activity elicited by c-Jun or CREB alone (191). This suggests that the formation of transactivating complexes on the STARD1 promoter is disrupted by overexpression of AP-1 proteins and CREB (185, 186, 191). This may be due to competition for limiting levels of the cofactor CREB binding protein (CBP) (191). Thus, AP-1 proteins may regulate STARD1 gene expression in Leydig cells basally at the CRE2/AP-1 site and support its response to cAMP, while CREB helps determine its cAMP sensitivity.
AP-1 Factors and Their Role in STARD1 Gene Regulation in Granulosa and Adrenocortical Cells.
In rat granulosa cells, the CRE2/AP-1 element of the murine promoter appears to only bind CREB. Post-luteinization, however, Fra-2 supplants CREB, binding the −81 to −75 site in rat granulosaluteal cells to foster cAMP-independent transcription of the STARD1 gene (116). Expression of a dominant Fra2/Fos negative protein inhibits promoter activity in luteal but not in less differentiated granulosa cells (116). This shift in binding partners reflects a change in transcriptional regulation that occurs with granulosa cell differentiation.
In primary bovine adrenal glomerulosa cells, AII increases c-Fos expression and c-Fos and c-Jun binding to the −95 to −68 region of the bovine gene which encompasses a putative AP-1 site (192). Overexpression of a dominant negative Fos suppresses basal and AII-stimulated STARD1 expression. In adrenocortical cells, c-Fos and Jun family members rise in response to AII and ACTH (193). In fasciculata-like Y1 cells, the rodent promoter exhibits a similar pattern of basal responsiveness to AP-1 proteins to that in MA-10 cells (188). As well, Jun proteins inflate the size of the cAMP response by rodent promoters, whereas Fos family members excluding FosB are generally inhibitory (114, 188). For instance, c-Fos dose-dependently inhibits basal and cAMP-stimulated rat promoter activity (114). Expression of both c-Fos and c-Jun attenuates the increase in basal and stimulated activity generated by c-Jun alone on the rat promoter (114).
One study in Y1 cells found that mutation of the CRE2/AP-1 site does not alter c-Fos inhibition of the rat STARD1 promoter, but when two other upstream sites at −1561 to −1555 and −187 to −181 are also mutated, basal and cAMP-stimulated activity increases (114). Experiments in clonal cells further suggest that c-Fos utilizes these sites to oppose SF-1 and repress promoter activity (114).
Overall, two interesting themes emerge from these studies. First, occupancy of the CRE2/AP-1 can change with cAMP stimulus or luteinization. Second, AP-1 proteins appear to have a support role in the cAMP response independent of their binding to this site.
Sterol Regulatory Element Binding Protein (SREBP).
Sterol regulatory element binding proteins (SREBPs) are also implicated in STARD1 promoter regulation. Under conditions of sterol-depletion, SREBPs are cleaved and migrate to the nucleus where they promote the transcription of genes involved in sterol availability, cholesterol synthesis and lipogenesis through the binding of sterol response elements (SREs) and coactivators like nuclear transcription factor Y (NF-Y) (194). Oddly enough, overexpression of SREBP-1a and variably SREBP-2 in human granulosa-derived or clonal cells increases promoter activity for sterol-depleting STARD1 (93, 195–198). Stronger induction is seen with SREBP-1c in nonsteroidogenic cells (197) and SREBP1 overproduction augments the response to (Bu)2cAMP with or without IGF-1 in mLTC-1 cells (53).
Several putative SREs lie in the distal rat STARD1 promoter, while others that bind recombinant SREBP-1a localize within −81 to −70 and −87 to −79 of the proximal promoter in the human and rat, respectively, overlapping the C3 C/EBP sequence (195, 196). Recombinant Sp1, NF-Y or SF-1 enhances binding of recombinant SREBP-1 to the rat SRE at −740 to −731 in EMSAs (197), but only NF-Y and SF-1 cooperate with SREBP-1a to facilitate rat promoter activity in nonsteroidogenic cells. The mouse promoter contains a smaller putative SRE four bp downstream of the GATA site. Mutation of this sequence decreases the magnitude of basal and EGF-stimulated promoter activity; however, SREBP-1 binding to this site has not yet been proven (55).
While these data are promising, endogenous SREBP regulation has not been demonstrated under conditions that promote SREBP cleavage. Addition of SREBP-inhibitory oxysterols to serum-starved Y1 cells fails to alter reporter gene expression but does elevate STARD1 mRNA levels in MA-10 and Y1 cells (48). While the addition of 27-hydroxycholesterol inhibits SREBP-induced LDL receptor promoter activity in human granulosa-derived and COS-1 cells cultured in delipidated media, STARD1 promoter activity again remains unchanged (195). These data question whether physiological conditions exist under which SREBP proteins do transactivate the STARD1 gene.
Liver X Receptor (LXR) and Retinoid X/Retinoic Acid Receptors (RXR/RAR).
The generation of oxysterols by StAR through its transfer of cholesterol to mitochondrial 27-hydroxylase and P450scc offers one means of feedback inhibition through oxysterol activation of LXRs. LXRβ (NR1H2) and to a lesser extent LXRα (NR1H3) are expressed in the adrenals and ovaries and an LXR response element (LXRE) is present at −200 to −185 in the mouse STARD1 promoter (199, 200). ChIP analysis using adrenal glands from various LXR-knockout mice indicate that LXRα is the predominant form that binds to the LXRE region (200). Stimulation of glucocorticoid synthesis by dexamethasone pretreatment of knockout mice increases adrenal STARD1 levels relative to wild-type animals. As well, reduction of both LXRs by RNAi stimulates STARD1 and SF-1 mRNA levels in H295R cells (199). A synthetic agonist of LXR inhibits steroid production by these cells as well as STARD1 and SF-1 mRNA basally, but has no effect on these genes in the presence of forskolin (199). However, the use of an LXR agonist in wild-type mice increases STARD1 expression in the adrenal gland (200); this effect may be secondary to LXR induction of ACTH generation and secretion by the pituitary (199).
In serum-free media, mutagenesis studies support a role for the LXRE in the induction of the STARD1 promoter (200). In these experiments, shorter term stimulation with a different LXR agonist or an RXR agonist increases STARD1 mRNA expression in Y1 and H295R cells (200). This stimulation is increased in the presence of both agonists. As might be predicted, supershift assays indicate that the LXRE is bound by recombinant LXRα/RXRα heterodimers (200). Thus, the nature of STARD1 regulation by LXRs may depend on the presence of serum and ligand.
9-cis-retinoic acid also upregulates STARD1 mRNA in K28 cells and increases promoter activity in K28 and Y1 cells (60). Specific retinoids also drive STARD1 gene expression and promoter activity in human theca cells (201). The action of retinoids in this case may involve a yet uncharacterized consensus retinoic acid response element (RARE) or potentially RAR or RXR heterodimerization with known regulators.
Yin Yang 1 (YY1).
The STARD1 gene promoter is also impacted by the conditional activator/repressor YY1. Recombinant YY1 binds an unconserved site within the −90 to −65 region of the human promoter that overlaps the proximal SREs (195). Selective mutation of the YY1 element enhances SREBP-1a-mediated STARD1 promoter activation in COS-1 cells (195). In rat luteal cells, YY1 associates with an upstream site within −1382 to −1193 bp to inhibit rat promoter activity (202). Increases in YY1 levels elicited by PGF2α correspond with a decline in STARD1 mRNA in rat ovaries (157, 196) and as would be predicted, PGF2α promotes YY1 binding to the upstream site (202). Targeted knockdown of YY1 reverses PGF2α inhibition of STARD1 expression (202).
While YY1 can inhibit SREBP-1a and NF-Y binding and activation of the rat promoter in clonal cells (196), it is unclear that YY1 and SREBP-1a directly interact (202). Instead, silencing of the STARD1 gene likely results from the recruitment of histone deacetylase 1 (HDAC1) to the promoter by YY1 (202).
Chicken Ovalbumin Upstream Promoter-Transcription Factor (COUP-TF) Family.
Other suspected repressors of the STARD1 gene include COUP-TF proteins. COUP-TF is decreased by AII in primary bovine adrenal glomerulosa cells coincident with an increase in STARD1 and aldosterone production (203). Exogenous expression of either COUP-TFI (NR2F1) or COUP-TFII (NR2F2) inhibits AII induction of the STARD1 gene and aldosterone synthesis as well as activation of the human promoter. Their site of action may reside within the proximal promoter. In glomerulosa nuclear extracts COUP-TFI binds the −49 to −26 bp region of the bovine STARD1 promoter in EMSA and this binding declines with AII stimulation (203). While the nucleotides for COUP-TF action have yet to be identified, this transcription factor family is known to recognize SF-1-like sequences (e.g., (204, 205)) and the −49 to −26 region does contain a putative SF-1 site. Moreover, using H295R nuclear extracts both COUP-TF and SF-1 interact with a human promoter fragment housing the −42 to −35 SF-1 site (163). Thus, COUP-TF could conceivably inhibit STARD1 gene transcription through competition with SF-1 for the proximal SF-1 site.
Other Mediators of STARD1 Transcription.
Other transcription factors implicated in STARD1 gene regulation include the ubiquitous transcription factor Sp1, which stimulates the human promoter in Y1 cells and cooperates with SF-1 (206). Sp1 also augments basal and cAMP stimulation of the murine promoter in mLTC-1 cells (53). Mutation of the Sp1 site at −146 to −137 reduces the magnitude of basal and EGF-stimulated promoter activity (55). On the other hand, Sp3 represses basal activity through the −180 and −150 bp region of the murine promoter in MA-10 cells via recruitment of HDACs (207). Forkhead box protein FOXL2 also represses basal activation of the human promoter in a clonal cell line (208).
Exogenous production of Kruppel-like factors (KLFs) 4, 9 and 13 suppresses basal expression of a STARD1-promoter reporter construct in luteinized porcine granulosa cells (209). Interestingly, lentiviral expression of KLF13 in the same cells stimulates endogenous STARD1 mRNA levels, leaving the role of this factor in STARD1 gene regulation unclear.
Regulation of STARD1 in the Rodent Placenta.
Few studies have examined the regulation of STARD1 in the giant trophoblast cells of the rodent placenta. In the absence of NR5A expression in the placenta, GATA2 and possibly GATA3 figure prominently (168). The murine GATA site binds GATA2 and participates in cAMP-stimulated STARD1 promoter activity in cultured cells (116). As well, the murine CRE2 element binds endogenous CREB-1 and ATF-2 to confer cAMP-responsiveness to the STARD1 promoter (116). Selective mutation of the C3 site indicates that C/EBP proteins are uninvolved.
Conclusions.
Numerous transcription factors participate in STARD1 promoter activation. When studies are evaluated by cell type some general patterns emerge. In cultured granulosa cells, gonadotropin-stimulated STARD1 promoter activity is primarily regulated by SF-1/LRH-1, GATA4/6, C/EBPβ and CREB. In rodent luteal cells, CREB activity appears to be replaced by Fra-2 to confer cAMP-independent activation. In Leydig cells, cAMP-regulated STARD1 promoter activity primarily involves SF-1, CREB/CREM, GATA4, and is modulated by AP-1 factors. In the adrenal cortex, SF-1, GATA6, AP-1 factors and CREM control STARD1 expression. Rodent placental giant trophoblast cells exhibit cAMP-dependent STARD1 regulation by GATA2 and CREB. Transactivation of STARD1 is inhibited by DAX-1, YY1, COUP-TF and conditionally by some AP-1 family members. Data with LXRs has been ambiguous, suggesting that the cellular setting for these factors is critical to their impact on STARD1. Regulation of the promoter by AP-1 proteins in non-rodent species remains unclear.
Hormonal Control of CYP11A1 Gene Expression
Regulation of CYP11A1 mRNA in the Adrenal Cortex.
Like STARD1, many serum and locally derived factors that increase steroidogenesis also increase CYP11A1 mRNA expression such as ACTH, AII, activin and BMP-6, albeit more slowly (41, 210, 211). Activation of PKC and Ca2+ signaling pathways, however, repress its transactivation (212). Stimulation of H295 cells with 8-bromo-cAMP increases the rate of transcription of the CYP11A1 gene and level of its mRNA (213). In human fetal adrenal cells, CRH, ACTH and cAMP analogs increase CYP11A1 expression (214, 215). Cyclic AMP also regulates promoter activity in Y1 cells (212). In adrenal cells, insulin and IGF-I by themselves have little effect on CYP11A1 mRNA levels or promoter activity, but potentiate increases induced by ACTH and cAMP (216, 217). Changes in aldosterone production with salt intake in rats correlate with CYP11A1 mRNA levels in the zona glomerulosa (218, 219).
Factors that inhibit adrenal steroidogenesis can also affect CYP11A1 gene expression. For instance, ANP and leptin oppose the rise in CYP11A1 mRNA elicited by ACTH in bovine adrenocortical cells (220, 221). TGFβ also inhibits CYP11A1 mRNA accumulation in fetal ovine adrenal cells (222).
Regulation of CYP11A1 mRNA in the Testis and Ovary.
Gonadotropins are the primary regulators of CYP11A1 expression in adult gonads. Gonadotropins and cAMP stimulate CYP11A1 promoter activity in granulosa, theca, luteal and Leydig cells (223–226). In Leydig cells, CYP11A1 is expressed at high levels basally and increases more slowly than STARD1 in response to cAMP, LH or hCG (7, 9, 227–230). Other hormones and growth factors that positively modulate CYP11A1 mRNA include thyroid hormone (58) and TGFβ in immature rat Leydig cells (62). Similar to their effects on STARD1 and steroid synthesis, IGF-I (52, 53, 231) and insulin (231) can stimulate CYP11A1 mRNA in certain Leydig cell lines depending on culture conditions. Negative regulators which may be derived from serum or be locally produced by Leydig cells or immune cells, include glucocorticoids (232), TGFβ (233), TNFα (67, 234, 235), interferon γ (236), leptin (66) and IL-1 (237). Activin A also reportedly lowers P450scc enzymatic activity in immature porcine Leydig cells (238). Thus numerous endocrine, paracrine and autocrine modulators contribute to the Leydig cell expression of CYP11A1 and steroidogenesis.
In contrast to StAR, P450scc is absent in Sertoli cells. However, it does appear in cultured immature cells with chronic stimulation by FSH or a cAMP analogue (74).
Like STARD1, expression of CYP11A1 in the theca cells of developing follicles in the ovary is driven by LH (76, 77, 239–241). While STARD1 mRNA is virtually undetectable in the granulosa cells of the pre-surge follicle of most species, CYP11A1 mRNA appears after antrum formation and rises with follicular development (77, 83, 240). CYP11A1 expression is further enhanced by the ovulatory gonadotropin surge and is maintained at high levels in luteal cells (240).
In cultured granulosa cells, CYP11A1 is induced by both FSH and LH (or analogue hCG), depending on the state of luteinization (89, 240, 242–244). There is a fair consensus that LH is needed to initiate the increase in luteal CYP11A1 mRNA in vivo. Depending on the species, continued expression of CYP11A1 during the luteal phase either remains reliant on LH or becomes constitutive (240, 245). Maintenance of luteal CYP11A1 during pregnancy depends on rescue of the corpus luteum by factors such as hCG in humans (246) and prolactin in rodents (164, 247). PGE2 also stimulates CYP11A1 mRNA accumulation in bovine luteal cells (112).
Gonadotropin stimulation of CYP11A1 expression is augmented by growth factors, such as IGF-II (92) and EGF (91, 97) in granulosa cells. One growth factor, IGF-I acts in a species-specific fashion in granulosa cells. In rodents, IGF-I alone has no effect on CYP11A1 in cultured granulosa cells, but can enhance the effects of gonadotropin (248). In the pig, IGF-I can act in concert with gonadotropin or by itself to induce granulosa expression of the enzyme (249, 250). IGF-I also upregulates CYP11A1 promoter activity in rat theca cells (96, 241).
Other positive regulators of both steroidogenesis and CYP11A1 include estradiol (240, 242, 244), progesterone (251), growth hormone (252), insulin (243, 253) and activin A (254) in granulosa, amphiregulin in cumulus-oocyte complexes (95), and insulin (253) in theca cells.
Consistent with their observed effect on steroidogenesis, negative regulators include PGF2α (255) and TNFα (256) in granulosa cells, GDF-9 in 8-bromo-cAMP-stimulated human granulosa cells (111), GDF-9 in theca cells (257), and PGF2α in luteal cells (258). TGF-β decreases CYP11A1 mRNA in cultured bovine granulosa cells (100), but increases P450scc protein in rat granulosa cells in the presence of FSH (101). In granulosa cells, several BMPs negatively regulate FSH-stimulated CYP11A1 mRNA or protein levels including BMP-4 (103), −6 (105) and −15 (108). The inhibitory effects of BMPs in these studies may be secondary to impaired FSH signaling (103, 105, 108). BMP-4, −6 and −7 also decrease basal CYP11A1 mRNA in theca cells (109).
Regulation of CYP11A1 mRNA in the Placenta.
Analogues of cAMP stimulate CYP11A1 mRNA and progesterone accumulation in primary cultures of human placental tissue and differentiating cytotrophoblasts (259–261) and human choriocarcinoma JEG-3 (262, 263) and BeWo cells (264, 265). The extracellular factors that propagate cAMP signals in placental cells in vivo, however, remain to be discovered. Activation of PKC also stimulates CYP11A1 expression to a lesser extent in JEG-3 cells (266). EGF which promotes cytotrophoblast differentiation can initiate PKC signaling (267) and weakly induce CYP11A1 mRNA (266, 268). Estradiol and progesterone increase CYP11A1 mRNA in cultured human syncytiotrophoblasts (269, 270), suggesting a positive feedback mechanism from placental steroids.
Unlike in the human, rodent placental CYP11A1 gene regulation is cAMP independent. Neither CYP11A1 nor progesterone output in cultured murine giant trophoblast cells is affected by cAMP (117, 118, 271). Differentiation as examined in Rcho-1 rat choriocarcinoma cells to the giant trophoblast-like phenotype is one mechanism that does induce CYP11A1 expression (272).
Transcriptional Control of the CYP11A1 Gene
CYP11A1 promoter activation involves trans regulatory factors common to the STARD1 gene as well as its own distinct regulatory factors. As with STARD1, the CYP11A1 gene is cAMP-responsive in most tissues. This responsiveness is concentrated in two areas of the human 5′-flanking region—one located within the first −120 bp and encompassing a fairly well-conserved region (Fig. 3) (273) and a second, unconserved sequence between −1633 and −1553 that participates in ACTH-stimulated promoter activity, as observed in transgenic mice (274). In addition to these cAMP response sequences (CRS) are other tissue- and species-specific enhancer and repressor elements. On the other hand, the placentas of human and mice have a completely separate mechanism of control independent of cAMP and SF-1. The following section summarizes the major cis- and trans-acting elements involved in CYP11A1 gene regulation.
SF-1 and LRH-1 Family and AP-2.
The CYP11A1 promoter is regulated by SF-1 in the gonads and adrenals. Both SF-1 and LRH-1 drive the human CYP11A1 promoter in granulosa cells and a clonal cell type (135, 142, 275). SF-1 overexpression induces CYP11A1 mRNA in bovine theca cells (276) and the rat CYP11A1 promoter in K28 Leydig cells (67). Selective knockout of SF-1 causes a catastrophic loss of P450scc in the testes (127). Also, CYP11A1 expression is cut in half in mutant Y1 cells with a deficit in SF-1 activity (133).
The proximal CRS in the human contains a highly conserved recognition sequence between −46 to −38 that binds SF-1 in granulosa, luteal and adrenal cell extracts as well as recombinant LRH-1 (223, 225, 274, 275) and participates in CBP/p300 recruitment in H295 cells (277). However, added SF-1 only drives basal not cAMP-stimulated activity derived from a minimal −55 promoter fragment in transfected adrenal and COS-1 cell lines; instead, the neighboring TATA box directs the cAMP response (278). In rodent granulosa cells, forskolin activation of the rat promoter partly localizes to the first − 73 bp containing the −50 to −43 SF-1 element within a region designated SCC1 (223); deletion of a second SF-1 site at −79 to −71 within the SCC2 region has no effect. In MA-10 cells, mutation of both rat SF-1 sites reduces basal but minimally affects cAMP-stimulated promoter activity (226). In the bovine promoter, the proximal SF-1 site located within the −57 to −32 region is instrumental to basal and cAMP-mediated activity in luteal and Y1 cells (225, 279).
Mutation of the proximal SF-1 site in the human promoter eliminates expression of a lacZ fusion gene in transgenic mouse tissues, suggesting that it is required for CYP11A1 expression in vivo (280). However, when the equivalent proximal SF-1 site in the endogenous murine CYP11A1 gene is mutated, transgenic mice exhibit only reduced testicular and adrenal steroid levels and impaired sensitivity of adrenal steroid production in response to stress (281). Ovarian and placental expression of P450scc is normal and no defects in reproduction are evident. These studies suggest that either the proximal murine SF-1 site is not essential for CYP11A1 production or other elements are compensatory. A second unconserved SF-1 site in the human promoter resides in the upstream CRS at positions −1617 to −1609. Mutation of this site similarly decreases the ability of the human promoter to drive reporter gene expression in the testes and adrenal gland of transgenic mice but not the ovary (280). Still, a critical role for NR5A proteins is indicated in the ovary since a granulosa-targeted knockout of LRH-1 dramatically reduces CYP11A1 mRNA in granulosa cells of hCG-treated mice (134).
Overall, NR5A proteins contribute to CYP11A1 gene regulation in steroidogenic tissues with an important exception—the placenta. SF-1 is absent in the murine placenta and while it is expressed in the human, it is apparently uninvolved. The proximal SF-1 site in the rat as in the mouse overlaps with an AP-2 binding site that does support promoter activity in murine placental giant trophoblast cells (282). Experimental data currently suggest that AP-2γ in the mouse and AP-2α in the human are more relevant to the control of CYP11A1 expression in this tissue (282, 283).
Long Terminal Repeat Binding Proteins (LBPs).
Expression of the CYP11A1 gene may also be influenced in human syncytiotrophoblasts by the long terminal repeat binding proteins LBP-1b, LBP-9 and LBP-32/MGR (284). These proteins bind to the placenta-specific − 155 to −131 bp region of the human promoter (285, 286). LBP-1b activates the CYP11A1 promoter in JEG-3 cells, whereas the cAMP-responsive LBP-9 may serve to modulate LBP-1b activity (287). LBP-32/MGR represses promoter activity in this choriocarcinoma cell line (285). An unrelated protein, transcriptional regulating protein of 132 kDa (TReP-132; transcriptional regulating factor 1, TRERF1), binds the same region as LBPs and drives human CYP11A1 promoter activity in both JEG-3 cells and H295 cells (287, 288). This factor also cooperates with SF-1 to facilitate CYP11A1 transcription in adrenal cells (289).
GATA, CREB and AP-1.
Overexpression of GA-TA4 or GATA6 can drive the human CYP11A1 promoter in COS-1, HeLa and adrenal cells (167, 290, 291). While computer analysis of the first 1000 bp of the 5′-flanking region of the CYP11A1 gene of humans, cows, pigs, rats and mice has revealed several putative GATA sites, their locations and number are not conserved (not shown). Only one study using the mouse promoter has characterized GATA binding and its function in a cell-specific context. The site located at −475 to −470 binds GATA4 in nuclear extracts of rat granulosa cells and GATA2 in mouse giant trophoblast cells (271). The GATA site participates in FSH activation of the promoter in granulosa cells and in cAMP-independent promoter activity in giant trophoblast cells.
Just downstream of this site resides a multi-functional CRE half-site at −450 to −447 (271). Similar to the murine STARD1 CRE2/AP-1 element, the CYP11A1 half-site binds CREB-1 in less differentiated rat granulosa cells and Fra-2 with more differentiation. The CRE half-site also binds CREB-1 in murine giant trophoblast cells, contributing to basal activity.
PMA-induced increases in CYP11A1 correlate with increases in c-Jun (189). An AP-1 binding site at −92 to −77 of the ovine CYP11A1 promoter mediates EGF and c-Jun activation in placental JEG-3 cells (292). The U-CRS region (−1633 to −1553) of the human promoter in Y1 adrenocortical cells is stimulated by c-Jun alone or in combination with c-Fos, whereas overexpression of a dominant negative Fos blocks c-Jun-mediated transactivation (274). c-Jun cooperates with SF-1 when both factors are overexpressed in JEG-3 cells to drive promoter activity through the U-CRS region (274) or via protein-protein interactions with SF-1 to activate a minimal 55 bp promoter construct containing the most proximal SF-1 site and the TATA box (293).
DAX-1.
Overexpression of DAX-1 in Y1 cells decreases endogenous CYP11A1 expression and forskolin-stimulated human promoter activity (159). In granulosa cells, co-transfection of DAX-1 opposes LRH-1 stimulation of the human CYP11A1 promoter (275). DAX-1 also blocks the ability of GATA-6 and SF-1 to drive the human promoter in nonsteroidogenic cells (167).
Sp1 and Polypyrimidine Tract-Binding Protein-Associated Splicing Factor (PSF).
The proximal region of the CYP11A1 promoter is G-rich and contains one or more Sp1 sites. The −118 to −100 region of the bovine promoter binds Sp1 and/or Sp3 in luteal, Y1 and H295R cell extracts (279, 294, 295). Mutagenesis studies show that this region cooperates with the proximal SF-1 site to drive basal and cAMP-stimulated promoter activity in both luteal and Y1 cells (279). A second binding site for Sp1 at −70 to −32 in the bovine promoter was also confirmed in Y1 cell extracts.
In porcine granulosa cells, forskolin and IGF-I independently activate the porcine CYP11A1 promoter through an IGF-I response element (IGFRE) located within the −130 to −100 region, which contains a functional Sp1 site (217). Sp1 binds to the 3′-end of the IGFRE and PSF binds to the 5′-end of the IGFRE (296). Overexpression of Sp1 increases basal CYP11A1 activity in granulosa and Y1 adrenocortical cells, whereas PSF overexpression decreases basal activity in both cell types and Sp1-driven activity in granulosa (296, 297). IGF-I does not activate the wild-type porcine promoter in Y1 cells; however, overexpression of Sp1 or mutation of the PSF element allows the promoter to respond to IGF-I (296). Taken together, PSF opposes the actions of Sp1 and controls IGF-I responsiveness of the promoter in different cell types.
An upstream enhancer region resides between −1898 and −1845 of the human CYP11A1 that contains two imperfect Sp1 sites, both capable of binding Sp1 in EMSA (298). Both sites participate in driving basal promoter activity in Y1 cells with the more proximal element, between −1875 and −1845, being the most potent. This site overlaps a NF-1-binding motif that also binds NF-1. However, NF-1 does not appear to be involved in CYP11A1 regulation.
Other Mediators of CYP11A1 Transcription.
Nur77 is implicated in increasing rat CYP11A1 gene expression in K28 Leydig cells, but has little effect on the promoter in human granulosa tumor cells (67, 142). High levels of KLFs 4, 9 and 13 in luteinized porcine granulosa cells suppress basal activity of a CYP11A1-promoter reporter construct (209). Additionally, lentiviral overexpression of KLF13 in these cells inhibits endogenous mRNA production, suggesting KLFs are repressors of basal CYP11A1 transcription.
As with STARD1, LXRs regulate CYP11A1 gene expression, but conflicting data exist in cell culture and knockout mouse models regarding whether it is inhibitory or stimulatory (199, 200). Incubation of human theca cells with specific retinoids increases CYP11A1 gene expression and promoter activity (201). As with STARD1, RXR/RARs may act at a putative RARE in the CYP11A1 gene or heterodimer with known existing regulators (201).
Conclusions.
Regulation of the CYP11A1 and STARD1 promoters has many similarities and some distinct differences. Basal and/or cAMP-mediated gonadal regulation of the CYP11A1 promoter involves SF-1/LRH-1, Sp1, GATA4, CREB-1 and AP-1 family members. Additional elements necessary for Leydig cell-specific expression of the mouse CYP11A1 promoter may reside 2.5–5 kb upstream of the transcriptional start site (9). Adrenal regulation involves SF-1, Sp1, GATA6, AP-1 and TReP-132. As with STARD1, DAX-1 and possibly LXR represses the promoter in adrenal and gonadal cells. Placental regulation of the gene is independent of SF-1 but rather exhibits cell- and species-specific regulation by AP-2 proteins, GATA2, CREB-1, AP-1 family members, LBPs and TReP-132. Unlike STARD1, roles for C/EBPs and NR4A members in CYP11A1 regulation are unclear.
Hormonal Control of HSD3B Gene Expression
Like CYP11A1, HSD3B gene expression rises more slowly than StAR with trophic stimuli in adrenocortical and Leydig cells. In the adrenal cortex, HSD3B is differentially regulated, paralleling changes in major steroid products between the zones.
Multiple genes exist for HSD3B in the human and the rodent. The major gonadal and adrenal transcript for 3β-HSD derives from the HSD3B2 gene in humans and the HSD3B1 gene in other species (3, 299). In this review, these forms of the gene will be referred to as HSD3B.
The 3β-HSD isoform in the placenta is the product of the HSD3B1 gene in the human and the Hsd3b6 gene in the rodent (formerly designated Hsd3b4 in rat), both hereafter referred to as placental HSD3B, although the latter enzyme also appears in postnatal Leydig cells in rodent (300, 301). Because the 5′-flanking regions of the adrenal/gonadal and placental 3β-HSD genes share little homology, their regulatory elements will be considered separately. Many of these elements are summarized in Figure 4.
Regulation of HSD3B mRNA in the Adrenal.
As mentioned before, the adrenal cortex expresses HSD3B throughout the rodent cortex, but not the human, where its expression is restricted to the outermost layers, the zonae glomerulosa and fasciculata (302). HSD3B mRNA rises in cultured human adrenocortical cells with known stimulators of steroidogenesis ACTH and AII (210). While ACTH stimulates HSD3B gene expression, cytokines TGFβ1 and LIF inhibit the accumulation of HSD3B transcripts in adrenocortical cells (28, 45, 303, 304).
Growth factors also stimulate HSD3B expression. For instance, EGF and IGF-I increase HSD3B mRNA levels in H295R (305) and bovine adrenocortical cells, respectively (28). Insulin, IGF-I and IGF-II increase HSD3B mRNA in cultured human adrenocortical cells (216, 306). Insulin and IGF-I also potentiate stimulation of HSD3B mRNA levels by ACTH and cAMP (216). Other factors stimulate HSD3B gene expression as well, including PMA and dexamethasone in H295R cells (305, 307) and CRH in cultured human fetal adrenal cells (214). On the other hand, TGFβ inhibits HSD3B mRNA accumulation in bovine adrenocortical cells (28).
Regulation of HSD3B mRNA in the Testis and Ovary.
The distribution of HSD3B is similar in many respects to CYP11A1 with some species variability. HSD3B mRNA and its protein are expressed in Leydig cells and the theca of developing follicles (9, 308, 309). 3β-HSD protein is absent in the granulosa layer of most species until late in follicle development, with the exception of the cow which expresses 3β-HSD protein much earlier (308, 309). HSD3B mRNA and its protein are further induced by the ovulatory gonadotropin surge and are produced in luteal cells (308, 310). In culture, LH, hCG and other cAMP-signaling agonists enhance HSD3B mRNA and protein levels in theca, granulosa, luteal and Leydig cells (227, 238, 245, 311–316). Activin A increases HSD3B enzymatic activity in immature porcine Leydig cells (238), while FSH stimulates HSD3B mRNA and protein in human and rodent granulosa cells (248, 317).
Insulin and IGF-I also increase HSD3B expression in granulosa cells (248, 317). While insulin cooperates with FSH to augment HSD3B mRNA in human granulosa cells (317), cooperation between IGF-I and FSH is not observed in rodent cells (248). In cultured theca cells from hypophysectomized immature rats, IGF-I increases HSD3B mRNA levels (316). However, insulin, IGF-I and IGF-II have no effect in human thecal tumor cells, though each do enhance forskolin-stimulated HSD3B mRNA expression (318). Several other growth factors have been shown to stimulate 3β-HSD protein or enzymatic activity in gonadal cells (319–322), but their effects on HSD3B mRNA have not yet been examined.
Luteotrophic concentrations of prolactin alone or combined with hCG increase HSD3B mRNA in rat luteal cells (238, 247). Levels of HSD3B also rise in luteal cells with PGE2 stimulation (112).
Negative regulators of HSD3B expression include PGF2α in granulosa and luteal cells (255, 323) and TNFα (324), dexamethasone (227), testosterone (227, 325) and IL-1 (324) in Leydig cells. A high luteolytic dose of prolactin inhibits HSD3B expression in rat luteal cells (326, 327). TGF-β negatively affects HSD3B mRNA in cultured bovine granulosa cells (100), but enhances FSH-stimulated 3β-HSD protein in rat granulosa cells (101).
BMP-2 (106), -4 (103), -6 (106) and -15 (108) decrease FSH-stimulated HSD3B mRNA or protein levels in granulosa cells, an effect associated with impaired FSH signaling (103, 106, 108). BMP-4, -6 and -7 also lower basal HSD3B mRNA in theca cells (109). Numerous other hormones and growth factors negatively impact 3β-HSD protein levels or enzymatic activity in gonadal cells including glucocorticoids (328–331), but their effects on HSD3B mRNA are not characterized.
The adrenal/gonadal HSD3B is the major isoform expressed in Leydig cells in the adult rodent testis and consequently, few studies have examined the regulation of the placental isoform (300, 301, 332). The murine promoter for placental HSD3B is sensitive to LH as observed in K28 cells, for instance ((67), personal communication, Dr. Keesook Lee, Chonnam National University, Gwangju, Korea). As well, TNFα inhibits placental HSD3B mRNA coincident with a reduction in testosterone production in rat R2C cells and knocks down LH-induced murine promoter activity in K28 cells (67). Knockout of the LH receptor in transgenic mice results in decreased levels of placental HSD3B mRNA in the testes without significant alterations in the signal for the adrenal/gonadal isoform (333), suggesting the former enzyme is more highly sensitive to LH. Since this isoform is homologous to the adrenal/gonadal protein (332), potentially some results of studies performed prior to the identification of the placental isoform in the rodent testis may actually reflect changes in placental HSD3B.
Regulation of HSD3B mRNA in the Placenta.
There are limited studies on placental regulation of HSD3B and the extracellular factors that are primarily responsible for its in vivo expression are not clear. Placental HSD3B mRNA expression is turned on in human cytotrophoblasts as they undergo spontaneous differentiation in culture. While HSD3B levels are enhanced by EGF during differentiation in the first day of culture, its spontaneous expression does not require the hormone (268). In JEG-3 cells, stimulation with a cAMP analogue or phorbol ester induces human HSD3B mRNA (334, 335). As with the CYP11A1 gene, estradiol and progesterone treatment of cultured human syncytiotrophoblasts increases human HSD3B mRNA expression (269).
Transcriptional Control of the HSD3B Gene in the Adrenal Cortex and Gonads
Less research has been devoted to the HSD3B promoter than STARD1 or CYP11A1 in part because the 5′-flanking regions of the mouse and human genes share little homology and those of many other species have yet to even be isolated. To date, almost all research has focused on the human promoter to delineate cis- and trans-acting elements as described below.
SF-1, LRH-1 and DAX-1.
One SF-1 binding site lies at −64 to −56 within a −101 to −52 bp stretch responsible for phorbol ester-driven promoter activity in H295R adrenocortical cells (336). This site is critical for SF-1-driven promoter activity and is responsible for PMA stimulation in HeLa cells. However, while SF-1 drives basal activity of the human promoter in human granulosa tumor cells and a clonal cell line (140, 142, 337), neither SF-1 nor LRH-1 stimulate promoter activity in MA-10 or H295R cells (146, 337). Mutation of the SF-1 site though eliminates 80% of basal activity in MA-10 cells, suggesting a role of NR5A proteins for HSD3B expression and perhaps the sufficiency of existing levels of these factors in this cell line (338).
LRH-1 and a cAMP analogue also additively increase human promoter activity in human granulosa tumor cells (140). This likely involves two additional NR5A sites at −906 to −900 and −315 to −309 that bind LRH-1 in extracts from this cell type and luteinized granulosa cells. Mutation of either site respectively blunts or eliminates the effect of LRH-1.
DAX-1 blocks LRH-1 induction of the promoter in granulosa tumor cells (140). Stable transfection of murine Y1 adrenocortical cells with DAX-1 opposes forskolin-stimulated HSD3B expression and human promoter activity (159). However, selective knockout of LRH-1 in granulosa cells has no significant effect on HSD3B expression in hCG-treated transgenic mice (134). Thus, the roles of NR5A family members in HSD3B promoter regulation may be limited.
NR4A Family.
The human HSD3B promoter also contains an NBRE at −133 to −125 (146, 338). Exogenous expression of Nur77 upregulates the HSD3B promoter in human granulosa tumor cells through this site, but is not synergistic with SF-1 (142). Overexpression of Nur77, Nurr1 or Nor1 stimulates promoter activity in MA-10 Leydig (338) and H295R adrenocortical cells (146, 338).
Nur77 may be more relevant to HSD3B function in the adult adrenal since it is expressed at approximately ten-fold higher levels than Nurr1 and Nor1 and cAMP stimulates its binding to the NBRE in supershift assays (146, 338). Comparatively, overproduction of Nur77 increases HSD3B levels in primary human adrenal cell cultures in excess of those elicited by ACTH (146). Mutation of the NBRE-1 site eliminates Nur77-stimulated activity in H295R cells (146).
In the human adrenal cortex, NR4A proteins like Nur77 may in fact determine zonal expression of HSD3B and thus the primary steroid produced by the adrenal cell—aldosterone/cortisol or DHEA (Fig. 1). Mirroring HSD3B expression, the level of these factors is highest in the glomerulosa and the fasciculata, but very low in the reticularis (146, 339). Such a role for NR4A proteins has been described for the gene for aldosterone synthase (CYP11B2) (145). An intriguing possibility is that as in the case of CYP11B2, SF-1 may countervail basal and AII-and Nur77-stimulated levels of the HSD3B gene (145).
One other factor, α-enolase, has been implicated in zonal HSD3B expression. The protein localizes in the nucleus of fasciculata but not reticularis cells and binds a specific site in HSD3B intron I (340). Through this site, α-enolase can stimulate HSD3B promoter activity in H295R cells.
GATA4 and GATA6.
While the 5′-flanking region of the human HSD3B promoter has at least four potential GATA binding sites at approximately −966, −570, −364 and −196, the most proximal site alone is enough to provide responsiveness to GATA factors (337). Both GATA4 and GATA6 are effective in H295R adrenocortical cells, whereas the promoter is especially responsive to GATA4 in MA-10 Leydig cells. While SF-1 and LRH-1 have little effect on promoter activity, co-expression of GATA4 or GATA6 synergizes with SF-1 and LRH-1 to increase reporter gene expression in both cell types (337).
Signal Transducer and Activator of Transcription 5 (Stat5).
A STAT5 consensus sequence also resides at positions −118 to −110 in the human HSD3B promoter. Prolactin is known to signal through the Janus kinase (Jak)/Stat pathway. While there are no studies on Stat5 control of the HSD3B promoter in steroidogenic cells, HSD3B promoter activity rises in HeLa cells co-transfected with the prolactin receptor in response to prolactin (341). The response is roughly doubled by co-transfection with Stat5 (341). Prolactin stimulates binding of Stat5 to the recognition sequence and mutation of this site eliminates promoter activation by the transcription factor. Stimulation of the promoter by EGF or dexamethasone also depends on the Stat5 site in HeLa cells (305, 307).
Farnesoid X Receptor (FXR).
The HSD3B gene may also be regulated by a different class of steroids, bile acids, acting through the nuclear receptor FXR (NR1H4). This receptor typically regulates genes involved in bile acid formation and lipoprotein and glucose homeostasis and is abundant in the adrenal cortex (342, 343). Recent microarray studies using adrenocortical cells identified HSD3B as a target of FXR (344). Overexpression of FXR stimulates HSD3B promoter constructs in H295R cells especially in the presence of a bile acid, while mutation of a FXR element at −137 to −124 blocks activation by this factor (344). Interestingly, glucocorticoids stimulate bile acid synthesis (345, 346). The possibility that bile acids through FXR in turn facilitate glucocorticoid production suggests that bile acids and glucocorticoids may coordinately regulate glucose metabolism (347, 348). As with STARD1 and CYP11A1, oxysterols may also regulate the HSD3B gene in the adrenal gland through LXRs (200).
Hypoxia Inducible Factor-1α (HIF-1α).
The testis constitutively expresses hypoxia inducible factor (HIF) 1α, part of a family of HIFs usually produced under hypoxic conditions to regulate genes needed to maintain tissue oxygenation (349). A putative HIF response element localizes to −1012 in the murine promoter and binds HIF-1α in EMSAs (350). Overexpression of HIF-1α drives murine HSD3B in TM3 cells, which are derived from immature Leydig cells.
Murine Placental HSD3B in Leydig Cells.
In K28 cells, overexpression of either SF-1 or Nur77 can drive the mouse placental HSD3B promoter (67). This transactivation is blunted by TNFα, which also inhibits stimulation of the promoter by LH. While these data indicate an important role for SF-1 and Nur77 in placental gene activation in the Leydig cell, regulation of the promoter is profoundly different in the placenta.
Transcriptional Control of the HSD3B Gene in the Placenta
Portions of the 5′-flanking regions of the placental murine and human HSD3B genes have been examined in transfection assays using human JEG-3 placental cells. SF-1 has no effect on the murine proximal promoter (351). Consensus sequences for AP-2, GATA, Dlx3 and transcription enhancer factor (TEF) are found in both human and mouse promoters. These placenta-specific enhancer elements reside over 2400 bases upstream of the transcriptional start site (351, 352). Binding by AP-2γ, Dlx3 and putatively, a TEF family member to sites −2885 to −2876, −2848 to −2843 and between −2869 and −2857, respectively, account for placenta-specific activation of the murine promoter (351). In the human, the former two proteins are irrelevant to specifying placental expression. Instead, a GATA binding site at −2559 to −2554 and a TEF-5 site at −2563 to −2558 confer placenta-specific promoter activity (352). EMSAs indicate that TEF-5 whose expression rises with cytotrophoblast differentiation binds to the latter site, while supershift assays have failed to resolve whether GATA-2, GATA-3 or GATA-4 is the determining factor that binds the GATA site in the human promoter. Future studies should clarify how these proteins may be involved in the cAMP- and PMA-responsiveness of the gene (334).
Conclusions.
As with the STARD1 and CYP11A1 genes, SF-1 or LRH-1 and GATA4 or GATA6 foster and DAX-1 inhibits human HSD3B promoter activity in gonadal and adrenal cells. The STARD1 and human HSD3B promoter also share regulation by Nur77. One striking difference of the HSD3B gene results from the promoter’s responsiveness to Nur77 in granulosa cells and with adrenocortical cell differentiation as well as the variable impact of SF-1. Distinctively, Nur77 and α-enolase contribute to the zonal specific expression of HSD3B in the human adrenal. Another difference from STARD1 and CYP11A1 is the regulation of the human HSD3B gene by the activity of FXR and Stat5. The placental human and murine HSD3B genes have distinct mechanisms of regulation from those governing the adrenal and gonadal HSD3B genes and from each other, with the murine promoter being primarily regulated by AP-2γ and Dlx3, and the human by GATA and TEF5. Similarly, the murine CYP11A1 and placental HSD3B genes share regulation by AP-2γ in placental cells. Additional studies are needed to clarify cAMP and growth factor control of the murine adrenal/gonadal and placental HSD3B genes in Leydig cells.
Challenges and Prospects
The STARD1, CYP11A1 and HSD3B genes share several common promoter elements and each possesses a few unique elements that dictate tissue specificity and coordinate their response to steroidogenic stimuli. In most cases, these genes respond to the same hormones in a similar manner, though with differing sensitivities and time courses. Pituitary hormones like LH and ACTH may act in part through cAMP-signaling pathways to coordinate activation of NR5A and GATA family members at each gene’s promoter, which in turn helps stimulate transcription. As the number of transcriptional factors involved in the regulation of these genes proliferates, how intracellular signaling pathways coordinate with the assembly of these factors at the promoter becomes of greater importance (353). One tremendous advance has been in our growing understanding of the shift in transcription factors that occurs with luteinization in granulosa cells.
On the other hand, few studies have addressed how these steroidogenic genes are regulated in the placenta. Placental regulation of these genes varies greatly from the other tissues and among animals, reflecting in part the diversity of placentation and roles of placental steroids between species (354).
Transcription factors also recruit coregulators such as CBP/p300 (128, 186, 191), steroid receptor coactivator 1 (SRC-1) (355, 356), Friend of GATA 2 (FOG-2) (357), steroid receptor RNA activator (SRA) (358) and receptor interacting protein 140 (RIP 140) (359) and can alter chromatin remodeling through modifying histone acetylation status such as with YY1 (202) or Sp3 (207) on the STARD1 promoter. Phosphorylation status (e.g., (191)) and physical associations between factors like c-Jun or c-Fos and Nur77 (152), SF-1, GATA4, and C/EBPβ (188), which can be independent of DNA-binding, also play important roles in the regulation of steroidogenic gene expression. The roles of transcription factor post-translational modifications, protein-protein interactions, cofactor recruitment and chromatin modifications require further study.
Some data on the transcriptional regulation of these genes are conflicting, as is the case for SREBP and LXR with STARD1, indicating the sterol environment of the cell may be critical as to how these factors impact the promoter. Another problem is that several different factors have been proposed to recognize sites that overlap with each other or are identical, such as in the case of the murine STARD1 promoter. Whether, for instance, a C/EBPβ, CREB or AP-1 family member binds the murine CRE2/AP-1/C3 element may depend not only on the stimulus and the cell type, but also the conditions under which they are tested. Such issues lead to the question of what tools should be used to determine how these genes are regulated. Data that rely on overexpression experiments in steroidogenic cell types may minimize the relative importance of factors like SF-1 due to pre-existing levels of the protein or confound results through the effects of such overexpression on other factors, like the stimulation of DAX-1 expression by SF-1 (360, 361). However, promoter assays in nonsteroidogenic cells that show cAMP-responsiveness lead to questions of relevance. The nature of such transfection experiments can also reflect chronic regulation not acute—an important distinction in the case of STARD1 and in luteinizing cells if the differentiation state is altered.
Redundancies in the system further confound our understanding of promoter regulation. For instance, despite indications of the importance of Nur77 in HSD3B expression, Nur77-knockout mice have no obvious deficits in steroidogenesis (362). One explanation is that other NR4A proteins like Nurr1 can substitute for it. The further use of selective knockouts of relevant genes like SF-1 and promoter elements in transgenic mice in conjunction with ChIP assays are important steps in clarifying gene regulation. In the case where animal studies are still not practical, selective reduction of transcriptional regulators with RNAi or other means accompanied by re-expression studies in appropriate steroidogenic cell types are needed. The impact of such loss of function and gain of function experiments on the endogenous promoters in their native settings will provide valuable insight into the regulation of the STARD1, CYP11A1 and HSD3B genes.
Overview of steroidogenesis. Cholesterol provides the substrate for de novo steroidogenesis. StAR (steroidogenic acute regulatory protein) mediates cholesterol delivery to the inner mitochondrial membrane and P450scc (cholesterol side-chain cleavage enzyme) in the adrenal, gonads and rodent placenta, but not in the human placenta. Molecules and enzymes mediating each step are indicated in italics. Boxes indicate the major tissues for each reaction. Abbreviations include Preg (pregnenolone), Prog (progesterone), DOC (deoxycorticosterone), ZF (zona fasciculata), ZR (zona reticularis), 3β-HSD (3β-hydroxysteroid dehydrogenase/Δ5-Δ4 isomerase), P450c17α (17α-hydroxylase/17,20-lyase) and 17β-HSD (17β-hydroxysteroid dehydrogenase). Alignment of the proximal regions of the STARD1 gene promoter of several mammalian species. Alignment of the proximal regions of the CYP11A1 gene promoter of several mammalian species. A. Known elements of the human HSD3B2 and murine Hsd3b1 gene promoters proposed to participate in adrenal or gonadal regulation of transcription. B. Elements of the human HSD3B1 and murine Hsd3b6 genes shown to participate in placenta-specific promoter activation. N.b., the murine placenta-type Hsd3b6 gene, is also significantly expressed in postnatal mouse Leydig cells. See main text for details.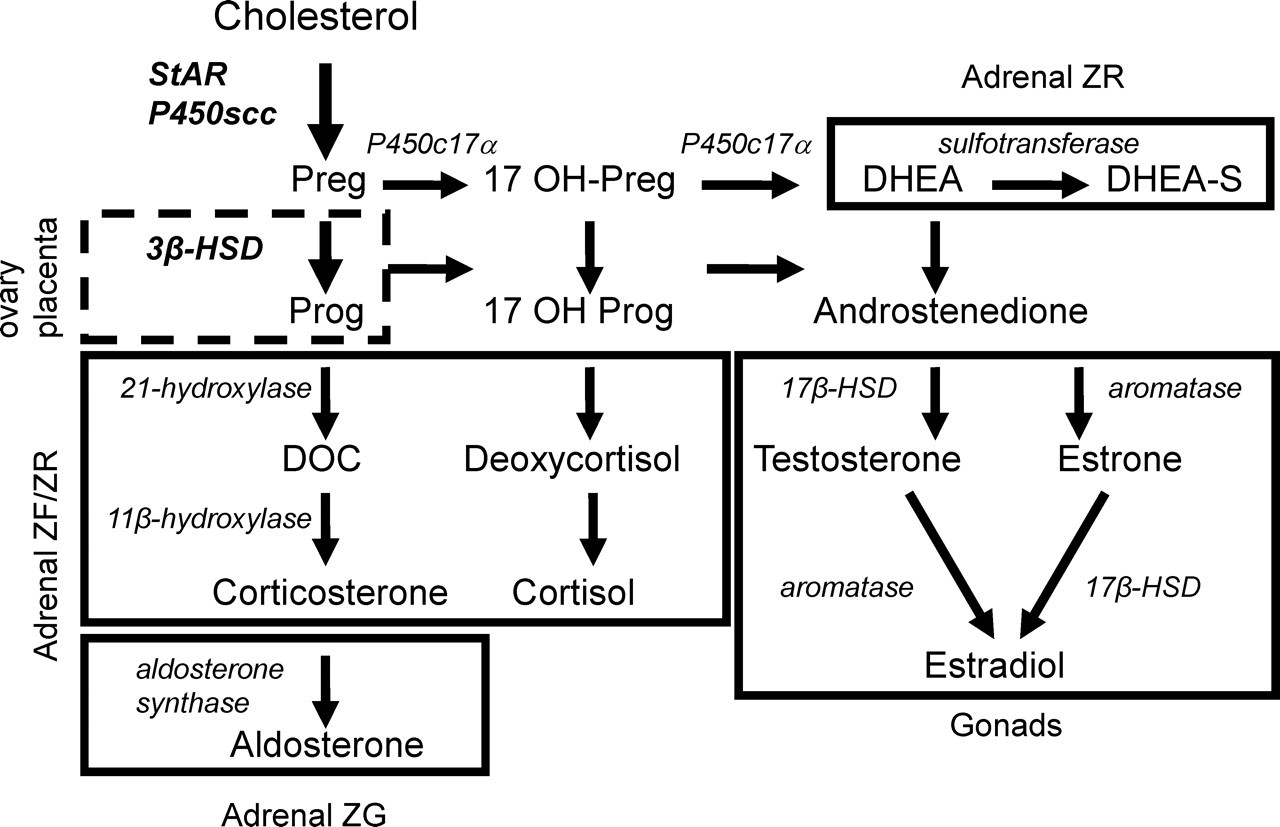
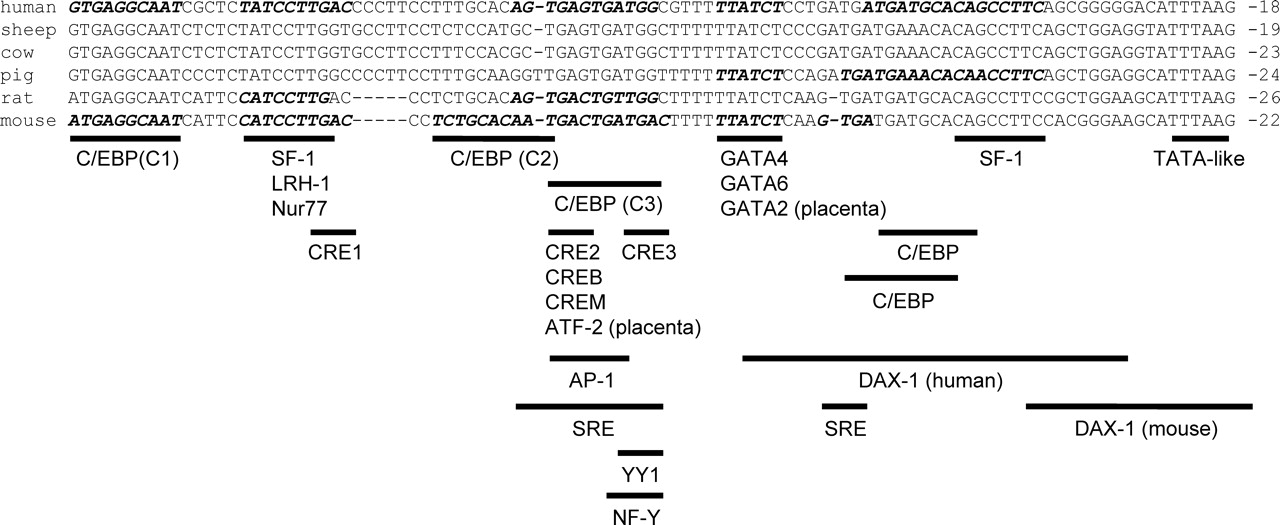
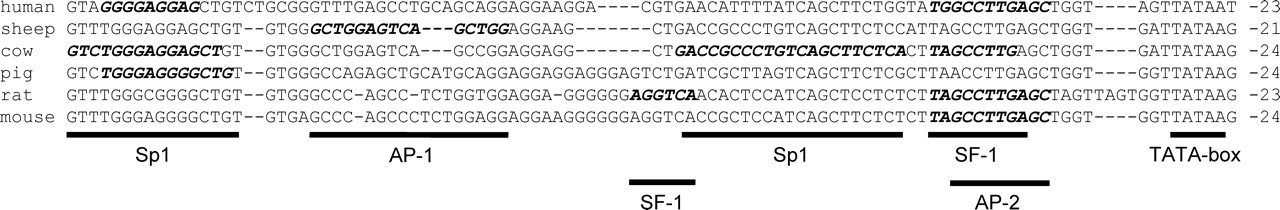

Footnotes
Acknowledgements
This article is dedicated to Dr. Anita H. Payne, who recently passed away. She was a pioneer in the field of reproduction and made major contributions to our understanding of transcriptional control of gonadal and placental steroidogenesis.