
Abstract
Modern hyperpolarization technology enhances the recordable magnetic resonance signal four to five orders of magnitude, making in vivo assessments of tracer pathways and metabolic compartments feasible. Existing hyperpolarization instrumentation and previous tracer studies using hydroxyethylpropionate (HEP) as an extracellular marker and 14-carbon label pyruvate as examples are described and reviewed as applicable to the working heart. Future metabolic imaging based on the use of hyperpolarized pyruvate needs to consider extra- and intra-cellular label dilution due to glycolysis, lactate oxidation and protein degradation. This dilution can substantially decrease the recordable signals from PDH flux (oxidative decarboxylation of pyruvate) and other pyruvate pathways. The review of previous literature and data suggests that the 13C-alanine signal is a better index of mitochondrially oxidized pyruvate than L-lactate. These facts and considerations will help in the interpretation of the in vivo recorded hyperpolarization signals of metabolic tracers and contrast media.
Introduction
In his Harvey Lecture, 1950, Dr. Richard Bing described the central role of the myocardial intermediary metabolism in health and disease (1). Magnetic resonance imaging (MRI) has had an important impact on diagnosis and understanding of cardiovascular diseases. However, the early promise of magnetic resonance spectroscopy (MRS), as a means of defining normal and disordered cardiac metabolism, has not yet been realized. Current practical problems due to low sensitivity and high costs of traditional magnetic resonance (MR) imaging hinder large-scale applications of this technology in both basic and clinical sciences as well as clinical diagnostics. The new technology of hyperpolarization may change this situation because it affects an increased signal to noise ratio by up to a factor of 105 at reasonable costs. Such high sensitivity is considered a promising “21st century approach” to the field of non-invasive metabolic imaging and mandates the development of non-toxic hyperpolarized contrast media.
Background—Why Hyperpolarization?
To achieve the goals of MR in the cardiovascular system, non-invasive imaging together with chemical identification greatly expand our ability to understand normal physiology and the perturbations which result from disease (2). Anatomy and dynamics of the heart are already well defined by proton MRI. 31P and 13C MRS have demonstrated the ability to identify energy rich metabolites, to record significant chemical changes known to underlie muscular contraction and to relate them to those dynamic activities (3). We are not confident that much more can be achieved, even with the increasing field strengths available to conventional MRI and MRS. MR is simply not sensitive enough—polarization is only a few parts per million, which translates into a relatively small number of nuclear ‘spins’ available for image reconstruction. This number is increased linearly with increasing field strength, so that 3 T ‘buys’ double the signal available at 1.5 T and so on. Conventional MR is also very slow—an examination may take 30 min or more. And, at a cost of approximately $1 million per Tesla as we move from 1.5 ($1–2 million) to 3 ($3–4 million) and then to 9.4 Tesla ($10 million) (4), conventional technology is simply beyond the reach of most modern health-care budgets and will certainly forever be restricted to advanced societies, and denied to the third world. At relatively low cost, hyperpolarization has the potential of solving many of these problems by amplifying MR signal 20,000–100,000-fold.
With this huge signal gain, hyperpolarization buys in vivo nanochemistry and metabolomics, decreases the time needed to observe a metabolite concentration change, better extracts the signal of a target molecule against a background of low spin species in a mixture, in vivo (i.e.13C background is only 1.1%, while water protons fall to zero in a hyperpolarized 13C imaging study) and permits studies to be performed faster at considerably lower cost. This last point is further enhanced by the likelihood that the spread of hyperpolarized MR, which is relatively insensitive to field strength, will reduce the rush to buy ever more powerful MRI scanners. We propose the umbrella term “High-Definition” magnetic resonance imaging (HD-MRI) to describe this technological advance.
Current Techniques and Preparation of Hyper-polarized Extracellular Markers (Contrast Media) and Rapidly Metabolized Metabolic Precursors
All of the techniques currently developed to achieve significantly greater polarization (enhanced ‘spins’ with P = 10–60% are equivalent to 20,000–100,000-fold signal enhancement) depend upon an apparatus termed a polarizer. A polarizer produces a deliverable contrast agent, which is then injected into a target Nuclear Magnetic Resonance (NMR) tube, an experimental animal or a human subject within an MRI scanner. Figures 1 and 2 illustrate four such polarizers, each of which operates on a different principle. Parahydrogen and Synthesis Allows Dramatically Enhanced Nuclear Alignment (PASADENA) is a liquid state chemical technique which operates at ‘room temperature’ giving rise to a liquid product (5) DNP—HyperSense is a solid state methodology of hyperpolarization by electrons and operates at liquid helium temperature with dissolution of the resultant frozen solid in warm water for injection of a liquid sample (6). High polarization in noble gases like 129Xenon and 3Helium was achieved within minutes to hours by optical pumping (7) in weak B field near ambient T. The polarized gases can be stored for ~8500 s at 77 K or 106 s at 4 K. The cost of photons (laser) is low and falling, thereby making this an inexpensive method of hyperpolarization. These hyperpolarized gases provide an inhaled hyperpolarized gas for inspiration or injection, and possibly employ the huge signal gain in 129Xe to 13C and 15N nuclei in biomolecules (8) while Quantum Relaxation Switch “Brute-Force” (9) is so called because it is also a solid state technique which employs very low temperatures and very high magnetic field with a liquid hyperpolarized product for injection (Kali-chefsky et al. unpublished work; Millikelvin Tech, MA).
PASADENA: Hyperpolarization Employing Para-hydrogen and Chemical Synthesis (5, 10).
Also known by a simpler name, Parahydrogen Induced Polarization (PHIP), the original discoverers selected the acronym “Parahydrogen and Synthesis Allows Dramatically Enhanced Nuclear Alignment”. Parahydrogen is a low energy spin state of hydrogen. By exposing native hydrogen (equilibrium mixture of 25% parahydrogen and 75% orthohydrogen) to iron oxide at a very low temperature, close to 100% parahydrogen conversion is achieved. Iron oxide (Hydrous Ferric oxide, commercially available as O-P Catalyst from Molecular Products, Inc., Lafayette, CO) acts as a catalyst by lowering the activation energy for the conversion of orthohydrogen to parahydrogen, thereby increasing the rate of this conversion. The heart of the PASADENA technique, however, is the chemical synthesis whereby the relatively short-lived hyperpolarized para-hydrogen spin is transferred in a symmetrical molecular addition to a third nucleus—in the case so far explored, 13C asymmetrically placed within a double bond to 12C, for which hyperpolarized state can last several tens of seconds (longitudinal relaxation times: T1), and equally to 15N. Both of these are non-radioactive stable isotopes of their respective nucleus. The spin physics required for efficient transfer of polarization is beyond the scope of the present review. Suffice to say that as new 13C double bond-contrast agents are discovered, new demands are made upon the spin order transfer sequence (Fig. 3). The initial transfer sequence (11, 12) and its improvements now provide the PASADENA technique with a broad range of biologically relevant, highly polarized (P > 40%) and non toxic contrast agents for cardiovascular applications, a few of which will be discussed below. The apparatus required (Fig. 1) is relatively simple and inexpensive with its heart being a very low field magnet, typically 1.75 MilliTesla, for the conduct of polarization transfer pulses, and a heated mixing chamber for the rhodium based catalyst which was found most effective in achieving symmetric molecular addition, a flowing source of parahydrogen and the 13C enriched reagent (13). Contrast reagents can be generated in injectable aliquots in rapid succession, because the chemical reaction and the spin transfer require only 3–4 seconds to reach completion.
Contrast Media
PASADENA Applications in the Cardiovascular System.
Cardiovascular applications for this metaboliz-able hyperpolarized reagent (Fig. 3) are anticipated and will be similar to those already demonstrated for DNP (see below). That succinate, while highly metabolizable once inside the cell and mitochondrial matrix, has a very limited cell membrane permeability relative to e.g. pyruvate, lactate, glucose, fatty acids or acetate. Thus, when succinate is applied externally through a catheter, only a very small fraction will enter intact cells. This is quite in contrast to metabolites such as glucose, pyruvate and lactate for which compounds powerful transporters exist on mammalian cell membranes. Free fatty acids (FFAs) also enter cells readily, not only via transporters but also due to their lipid solubility. Weak acids such as acetic acid or propionic acid also typically enter cells readily, since they are only weakly charged at physiological pH 7.4 and have some lipid solubility. Based on such considerations, we do not overemphasize the utility of hyperpolarized succinate in the context of metabolic imaging. On the other hand, succinate could perhaps be developed for extracellular imaging, of the coronary tree (exactly because of its poor membrane permeability and essential absence of extracel-lular metabolism). In the meantime, a novel, water-soluble PASADENA reagent which is not metabolized, 2-hydrox-yethyl propionate (HEP) has been employed in rodents (14), to track catheter positions, outline the contours of heart and lungs (Fig. 4) and, in pigs, to provide real-time coronary angiography (Fig. 5), catheter positioning and myocardial perfusion (15, 16). Petersson and colleagues make a strong case for hyperpolarized 13C real time imaging in the cardiovascular system and illustrate that with i) catheter tracking in real time and ii) quantification of myocardial perfusion. Catheter tracking is attractive because hyper-polarized 13C imaging can be performed against a zero background (Fig. 5A) and myocardial perfusion using hyperpolarized HEP (Fig. 5B) is an advance, since this can be quantitative and therefore, a major improvement upon the current Gd-based myocardial perfusion. The issue of i.v. toxicity of HEP remains to be investigated.
Design of a Plaque-Targeting PASADENA Reagent.
Our group has worked on a strategy that takes this well validated PASADENA reagent (HEP) into cardiovascular molecular imaging. Speculating that the PASADENA reagent HEP already described, could be targeted to any given tissue or receptor, simply by selecting the binding reagent (Fig. 6A), the molecule shown in Figure 6B was created (17). This molecule was almost as efficiently hyperpolarized by PASADENA (P ~25%) as was the ‘free’ HEP itself (P > 40%) and a subsecond 13C image of tetrafluoropropyl propionate (TFPP) was created after injection into a segment of isolated porcine aorta (Fig. 6C). T1 of the resulting hyperpolarized 13C MR signal was marginally shorter than the free HEP but still ample for in vivo imaging of this targeted PASADENA reagent. We have recently confirmed that hyperpolarized TFPP binds to an analogue of plaque, dimyristoyl palmitate (DMPC). Based on a small, but significant chemical shift difference from free reagent, TFPP has potential to become an in vivo plaque imaging reagent. Studies are now in progress using a murine hyperlipidemia model (courtesy of Dr. Wanda Reynolds, Sidney Kimmel Cancer Institute) to determine whether this can be applied to vulnerable plaque in vivo. More generally, we believe that an analogous strategy whereby a simple double-bond-13C configuration suitably hyperpolarized, is integrated chemically with a specific binding ligand and water-solubilizing moiety, will fuel a generation of designer-hyperpolarized molecular imaging reagents of the future.
Metabolic Imaging
DNP Applications in the Cardiovascular Systems.
Dynamic Nuclear Polarization (DNP) employs low temperature and high magnetic field to transfer polarization from electrons to nuclei (18) and was developed for biomedical use by Golman et al.; subsequently, GE Healthcare. A ‘bench-top’ version is also available from Oxford Biotools (see Fig. 2) and both have been used in cardiovascular applications. In each case, the 13C molecule employed was 1-13C pyruvate for metabolic imaging. Validation and analytical power of this technique have been demonstrated in pre-clinical studies. A growing range of applications of DNP exists in oncology (19). However, the future appears bright for this procedure in the cardiovascular system. Hyperpolarized 13C pyruvate is rapidly metabolized by many tissues, entering different pathways. In cancer models, where glycolysis predominates, a major proportion of the 13C hyperpolarization appears rapidly in the lactate pool reflecting intense reductive pyruvate metabolism. Hyperpolarized 13C lactate can thus be used to create an anatomic 13C MR image of the tumor (not shown here), against the very low background of normal tissues, with mainly oxidative pyruvate metabolism. The ability of hyperpolarized 13C MRS to qualitatively reflect flux through a single enzyme, in this case pyruvate dehydrogenase (PDH), was demonstrated in the non-working isolated rat Langendorff heart perfused with hyper-polarized 13C pyruvate at a concentration of 2 mM (Fig. 7). Metabolic flux through PDH was indexed by 13C bicarbonate production, while flux through the TCA cycle was derived from conventional 13C isotopomer analysis of glutamate in cardiac extracts. When PDH flux was inhibited by the medium-chain fatty acids octanoate and propionate (2 mM; caveat: 2 mM octanoate is a weak uncoupler (see below)), 13CO2 and H13CO3 production were reduced to below detectable levels, reflecting known negative feedback inhibition by β-oxidation of fatty acids of PDH flux and PDH activity (20–25). In each case, the point is made that hyperpolarized 13C MRS is a powerful technique with which to measure instantaneous metabolic adaptation in the intact heart. With careful experimental design and new hyperpolarized 13C probes, it will be possible to reach biologically insightful conclusions that dramatically impact our understanding of cardiac function.
The next illustration is in the context of myocardial ischemia/reperfusion resulting in either stunning (reversible non-necrotic injury) or myocardial infarction (myocardial cell death). In the in vivo porcine heart, i.v.-applied hyperpolarized 1-13C pyruvate is rapidly transported intra-cellularly and instantly metabolized to three major products: alanine (alanine aminotransferase, glutamate-pyruvate transaminase), L-lactate (lactate dehydrogenase) and bicarbonate (in the steady metabolic state about 90% of myocardial (bicarbonate + CO2) formation is due to oxidative decarboxylation of pyruvate by mitochondrial pyruvate dehydrogenase (PDH) (26–28) (Fig. 8A). Following zero-flow ischemia plus reperfusion (complete occlusion of the left circumflex coronary artery for 45 min followed by 120 min reperfusion), hyperpolarized pyruvate metabolism is altered dramatically (Fig. 8B). 13C images show the myocardial distribution of hyperpolarized pyruvate. The final injection solution in these trials contained 300 mM pyruvate, 100 mM TRIS buffer (equivalent to ~500 mOsm/L), 0.27 mM Na2 ethylenediaminetetraacetate (EDTA), 250 mM sodium ions and ~50 μM paramagnetic agent. The temperature and pH were approximately 30°C and 8.2, respectively. The pigs were intubated, and two needles were inserted: one in the hind leg for administration of anaesthetic agents and a second one in the front leg for hydration with Ringer solution. The second needle was also used for administration of 13C pyruvate contrast medium. The tracer was injected during reperfusion at 15 min or at 20 min and its immediate metabolites, CO2, bicarbonate, L-lactate and alanine in short axis views (which shows a single cross-sectioned slice through mid-ventricle) were recorded, each image requiring only 3 s. When comparing the upper panel of images (normal control) with the infarcted post ischemia myocardium (middle panel), it becomes obvious that in the infarcted area of the outer left ventricular wall there are no detectable bicarbonate and lactate signals, but a weak residual alanine signal still is recordable. On the other hand, the inner wall and septum still show normal signal intensities, as expected, compared to the control in the upper panel. These observations suggest that the infarcted (dead) myocardium has ceased to decarboxylate pyruvate, most likely because the PDH is inactivated (29) and the mitochondrial electron transport chain is probably damaged, rendering it unable to effectively oxidize NADH reducing equivalents and generate the proton gradient required for ATP synthesis. The resulting intra-cellular acidosis ultimately leads to cardiac cell death. Another potential explanation could be that the infarcted area is in a low-reflow state (ischemia) despite global reperfusion, an ischemic condition which would lead to intramitochondrial NADH accumulation and resulting in PDH flux product inhibition and PDH inactivation due to metabolic regulation of the interconvertible enzyme (23, 30–36). A low-reflow condition in the infarcted area could also explain the absence of a detectable L-lactate signal, which however could also reflect LDH washout in early reperfusion and/or the possibility that, while there is accumulation of cytosolic NADH in the low-flow ischemic zone, too little or no pyruvate tracer at all reached the infarcted area. On the other hand, the existence of a weak alanine signal from the infarct area argues against a complete zero-reflow condition.
The lower panel of Figure 8 shows the signals from hyperpolarized pyruvate in myocardial stunning. Stunned myocardium is obtained in reperfusion after a short 15 min period of myocardial ischemia; the reperfused ventricular myocardium is in a low-function, delayed-recovery state, a condition known as “stunned myocardium” (37–40); the stunned myocardium typically has no infarct and no necrotic myocytes, its contractile reserve is preserved and the hypodynamic state is temporary and usually fully reversible within hours (38, 41–44).
The first hyperpolarization studies to image stunned myocardium were published by Golman et al. (45) using i.v. [1-13C]pyruvate, injected at 2 hr after start of reperfusion. In the stunned myocardium the data showed near-normal L-lactate, alanine and bicarbonate signals (lower panel of Fig. 8) indicating essentially intact cytosolic and mitochondrial pyruvate metabolism; these novel hyperpolarized cardiac pyruvate data are consistent with earlier studies in isolated working perfused guinea pig heart stunned by reperfusion; here pyruvate (plus glucose) dose-dependently restored normal energetics and ventricular pressure development within about 30 min of reperfusion (44, 46).
Upon close re-inspection of the Golman et al. porcine STUNNING data compared to those from the their INFARCT protocol (Fig. 8 upper and lower panels), it seems obvious that the metabolic image from the stunned heart predicts the better outcome clinically, mainly because stunning did not disable oxidative mitochondrial pyruvate decarboxylation. On the other hand, the infarct protocol documented the absence of pyruvate oxidation, indicating the absence of mitochondrial respiration and thus pointing to failure of the vital mitochondrial oxidative phosphorylation. This interpretation is consistent with the already discussed transitory nature and full reversibility of myocardial stunning, typically not leading to cardiomyocyte death. Nevertheless, a small section of the stunned myocardium in Figure 8K failed to display the bicarbonate signal (outer wall of left ventricle) while the alanine or the L-lactate signals were well detectable. This observation could suggest a cell death area within the viable stunned area, since it indicates cessation of oxidative pyruvate decarboxylation, perhaps due to mitochondrial failure locally in this particular experiment. If this interpretation is correct, there must have been severely injured foci within the stunned myocardium that resulted in mitochondrial failure to oxidatively decarboxylate the [1-13C]pyruvate. In a clinical context, we suspect such forms of irreversible injury would manifest Gd-delayed enhancement in conventional cardiac MRI.
Key Determinants of Myocardial Pyruvate-Uptake and Oxidation in the Physiologically Performing Heart
The Petersson study requires consideration of the lessons now to be learned using hyperpolarized 13C cardiac imaging. The main elements we will consider here are: i) the concentration dependence of cardiac pyruvate uptake and oxidation at constant workload, ii) the effects of left ventricular work output at constant pyruvate concentration, iii) the effects of positive or negative inotropes, iv) the steady state kinetics of pyruvate uptake and oxidation based on special titration protocols and non-linear curve fitting, and v) the direct temporal relationships between altered work output or contractile state and rates of pyruvate oxidation. Pyruvate oxidation was indexed by 14CO2/ H14CO− 3 production from [1-14C]pyruvate. This review compiles published and new data and may provide useful, if not critical information for optimally designing and evaluating non-radioactive 13C-NMR metabolic signals based on hyperpolarized [1-13C]pyruvate technology in the physiologically performing heart (6, 47). In fact, a comprehensive understanding of the recent in vivo results from hyperpolarized 13C studies, can be obtained from earlier reported metabolic studies using radioactive 14C.
With respect to the metabolic condition of the heart in vivo, it is clear that the myocardium can function normally under a multitude of hormonal, inotropic, metabolic and pressure-volume-load conditions. The working heart can rapidly adjust or respond metabolically to changing demands of the myocardium itself as well as to those from the systemic circulation. To maintain the myocardial energy balance and hence normal ventricular function, anaerobic glycolysis is not sufficient. It is only the mitochondrial oxidative phosphorylation of ADP in the respiratory chain that is capable of maintaining cytosolic energetics at levels required for stable hemodynamics in the physiologically working myocardium. The respiratory chain receives reducing equivalents from the citric acid cycle and mainly from four other input pathways 1 : the malate-aspartate shuttle (transfer of NADH reducing equivalents from glycolysis and lactate dehydrogenase across the inner mitochondrial membrane) (48–52), the pyruvate dehydrogenase (intra-mitochondrial NADH and acetyl-CoA via oxidative decar-boxylation of pyruvate), the FFA oxidation cycle (β-oxidation) and the mitochondrial β-hydroxybutyrate dehydrogenase (oxidation of D-β-hydroxybutyrate) (Fig. 9). The pyruvate dehydrogenase system is a high-capacity multi-enzyme complex (22, 34, 36, 53–58) that is the main controller of the entry of carbohydrate and lactate carbon into the citric acid cycle. These major pathways enable the working heart to rapidly adjust to or take advantage of variable systemic metabolic conditions such as hyper-glycemia, lactatemia, ketonemia and high levels of fatty acid β-oxidation. Varying hemodynamic loads and catechol-amine stresses strongly influence the rate of glycolysis, pyruvate decarboxylation and β-oxidation of fatty acids. However, it is not clear how such variable metabolic/ hormonal/hemodynamic conditions can affect NMR recordable signals, if based on the hyperpolarized [1-13C]pyruvate technology. To address this experimentally we quantified to what extent altered systemic metabolic and cardiac hemodynamic states, both prior to and during adrenergic stimulation, influence cardiac pyruvate uptake and oxidative decarboxylation using the controlled setting of the isolated hemoglobin-free perfused but physiologically performing heart. We measured the active and inactive forms of the myocardial PDH and compared this data with 14CO2/ H14CO− 3 production from [1-14C]pyruvate in response to altered left ventricular preload (volume load) or afterload (pressure load), both in the absence and presence of physiological concentrations norepinephrine (NE, 0.02–0.50 μM ). The metabolic rate data correlated with key cardiac function parameters such as left ventricular pressure development and dP/dt, pressure-volume work and myocardial oxygen uptake.
The cardiac α-glycerophosphate cycle is not listed here, since it is a low-capacity system relative to the malate-aspartate shuttle. The cardiac α-glycerophosphate cycle cannot transfer sufficient cytosolic reducing equivalents to the mitochondrial matrix to maintain energetics and hemodynamic stability of the working heart (48).
To simulate the principle metabolic and adrenergic conditions of the heart in vivo, protocols were designed that examined pyruvate oxidation in the isolated working hearts subjected to the five main systemic metabolic states in the absence and presence of NE: normoglycemia, hypoglyce-mia, lactatemia, ketonemia and various levels of FFA β-oxidation. Our experimental model was the isolated working guinea pig heart, perfused in a non-recirculating system with a hemoglobin-free 95% oxygenated Krebs-Henseleit medium, pH 7.4, 37°C; this model was used as it displayed the Frank-Starling mechanism and coronary flow autoregulation in response to increased pressure loads; this model also showed increased work output and contractile state combined with coronary dilation in response to the physiological vasoconstrictor, norepinephrine (NE) (58–61). In the studies on PDH flux and PDH activity, the isolated hearts were perfused at constant pre- and afterloads while the NE concentration was varied over the physiological range, 0.02–0.5 μM. In this set of experiments hearts metabolized 1 mM [1-14C]pyruvate in presence of 2–5 mM D,L-β-hydroxybutyrate to downregulate the PDH system; this approach enabled us to quantify extent and direction of the adjustments of PDH flux or PDH activity in response to physiological adrenergic stress. In another set of experiments, special pyruvate titration protocols were designed to assess the steady state kinetics of pyruvate uptake and oxidation under the principle metabolic states; here arterial pyruvate concentrations were stepwise increased from about 0.1 mM to 2–4 mM at constant left ventricular pre- and afterloads, both in the absence and presence of constant concentrations of NE. The principle PDH-flux-modulating metabolic states were normoglycemia (alternative metabolic substrate 5 mM glucose (+ 5 U/L insulin)), severe hypoglycemia (pyruvate as sole substrate or 1 mM glucose as cosubstrate), normal and severe lactatemia (1–10 mM lactate as cosubstrate), severe ketonemia (2–10 mM D,L-β-hydroxybutyrate 2 as cosubstrate) and high levels of FFA β-oxidation (1 mM or 0.2 mM octanoate as cosubstrate). To understand this complex system further, we review our earlier data (20, 26, 58, 60, 62–64) and add some new data; the results are discussed relative to data from other laboratories in the field (20, 22, 23, 25, 27–36, 53, 55–57, 65–84) and in regard to their implications for the novel pyruvate-hyperpolarization technology in the heart pioneered by Golman and colleagues (6, 19, 45, 47, 85, 86).
DL-β-hydroxybutyrate was added to the arterial perfusion medium; 40–42% of the racemic DL-β-hydroxybutyrate was the metabolizable D-β-hydroxybutyrate.
Ventricular Work Output or Inotropic State Determines Cardiac Oxidative Pyruvate Fluxes.
Table 1 summarizes demand-induced changes in PDH activity and flux in the normoxic physiologically performing heart metabolizing pyruvate under simulated ketonemic conditions, both in the absence and presence of low and high physiological NE concentration (0.02–0.50 μM) to alter cardiac contractility over the physiological range. Isolated working guinea pig hearts were metabolizing 5 mM D,L-β-hydroxybutyrate in presence of 1 mM [1-14C]pyruvate. The data show that the PDH system was substantially down-regulated as the PDHa accounted for only ≈40% of PDHt and 14CO2/H14CO− 3 production, the measure of PDH flux, accounted for even less, ≈20% of PDHa or ≈7–9% of PDHt. Despite these substantial inhibitions of PDHa activity and flux due to ketonemia, 14CO2/H14CO− 3 production from [1-14C]pyruvate increased disproportionately as a function of the NE-concentration; the NE-induced increase in 14CO2/H14CO− 3 production was much larger (up to 7.1-fold) than the associated increases in ventricular mechanics or myocardial oxygen uptake: ventricular dP/dtmax increased 3.7-fold, work output 1.7-fold, heart rate 1.4-fold and the heart-rate-pressure product 1.5-fold. Under the same conditions, PDHa or MVO2 increased 2.4- or 2.1-fold, respectively. The maximum physiological NE concentration (0.50 μM) activated PDHa almost fully (90% of PDHt) but 14CO2/H14CO− 3 production remained inhibited (it was only 58% of PDHa) most likely due to the increase in mitochondrial NADH and acetyl-CoA due to the imposed severe ketonemia. These data suggest that acute cardiac PDH flux can be exceptionally responsive to the ventricular inotropic state or work output even when the PDH system is downregulated and highly inhibited due to ketonemia.
Working hearts were also examined in the absence of ketonemia using 1 mM pyruvate as the sole substrate; this protocol simulated a status of minimal downregulation of PDH (PDHa/PDHt ≈80%) and minimal PDH flux control by product inhibition (NADH, acetyl-CoA) (58, 60, 62–64). Results revealed the full potential of PDH flux adaptations in response to acutely altered physiological work states: When the left ventricular preload was changed at constant left ventricular afterload or vice versa, the changes in 14CO2/H14CO− 3 production, i.e. pyruvate oxidation, accounted for essentially 100% of the associated changes in myocardial oxygen uptake (58, 62). Clearly, in the well-oxygenated and physiologically working myocardium the conversion of exogenous [14-C]-labeled pyruvate to carbon-labeled (CO2+HCO3) can be highly reflective of altered cardiac work and inotropic states, and this is true even under conditions of substantial downregulation of the PDH system due to high levels of ketone bodies.
Transitory Minute-by-Minute Regulation of Cardiac 14CO2/H14CO− 3 Production from [1-14C]Pyruvate Under Various Ventricular Work and Metabolic States.
Normoglycemia.
Detailed time course studies also showed that 14CO2/H14CO− 3 from [1-14C]pyruvate faithfully tracked cardiac work output and inotropic state with an at least 1 min time resolution. This was obvious when acute demand-related changes in cardiac 14CO2/H14CO− 3 production were studied in transition states from normal contractility to increased contractility or vice versa. Tight temporal correlations between work output and pyruvate fluxes were observed in normoglycemia and ketonemia states. Figure 10 depicts the acute effects of NE wash in and wash out on 14CO2/H14CO− 3 production from [1-14C]pyruvate and on ventricular work output with a minute-by-minute resolution. Panel 10A shows the results from simulated normoglycemia, panels 10B, C those from severe ketonemia. In all conditions the arterial [1-14C]pyr-uvate concentrations were in the physiological range between 0.1–0.3 mM. In the normoglycemia of panel 10A both the 14CO2/H14CO− 3 production from [1-14C]pyruvate and work output approximately doubled within less than 2 min when NE infusion was started. In addition, both the transitory and steady state 14CO2/H14CO− 3 rates directly correlated with the changes in work output with at least a 1 min time resolution. When NE was washed out, a similarly tight correlation between decreasing work output and declining 14CO2/H14CO− 3 production was observed. Unlike the 14CO2/H14CO− 3 production, the lactate release showed no such parallel and tight temporal correlations with transitory or steady state changes in work output or contractility.
Ketonemia.
Figures 10B, C show results from similar protocols under the conditions of severe ketonemia, again in the presence of low physiological pyruvate concentrations. As expected the absolute rates of 14CO2/H14CO− 3 production from [1-14C]pyruvate were greatly reduced (Table 1) (23, 25, 58, 60, 63, 64), accounting for only ≈5–10% of the rate measured during normoglycemia in Figure 10A despite comparable work outputs. Despite this strong downregula-tion of pyruvate oxidation, the temporal synchronization between changing ventricular work output, NE positive inotropy and 14CO2 production was retained during ketonemia. The conditions of panel 10C simulate an extreme case of ketonemia relative to that of panel 10B; here the D,L-β-hydroxybutyrate concentration was doubled from 5 mM to 10 mM and the [1-14C]pyruvate concentration was decreased from 0.3 mM to 0.1 mM. NE infusion again disproportionately stimulated 14CO2/H14CO− 3 production (about 300%) along with work output (about 70%) and oxygen uptake (about 100%). When applying the L-type calcium channel blocker D600 (a verapamil derivative) during NE stimulation, we observed minute-by-minute parallel decreases in work output and 14CO2/H14CO− 3 production. Clearly, the Ca2+ channel blocker decreased work output and contractility and this negative inotropic effect was reflected immediately by PDH flux decreases. In addition, the D600 effects were largely reversible during the D600 washout phase. Close inspection of the oxygen uptake data from panel C data also reveals a close temporal association between 14CO2/H14CO− 3 production from [1-14C]pyruvate and myocardial oxygen uptake. Since myocardial oxygen uptake directly reflects mitochondrial respiration, the data show that the observed changes in PDH flux were driven by altered myocardial demand due to altered work output and inotropy due to NE with and without compound D600. It is remarkable that these PDH flux-work/inotropy correlations could be demonstrated even under the heavy metabolic constraints due to severe ketonemia or artificially high acetatemia and even when the arterial pyruvate level was in the low physiological range between 0.1–0.3 mM (58, 60, 62).
β-Oxidation.
What is the relationship between PDH flux-work/inotropy during myocardial FFA β-oxidation? Imposing high rates of fatty acid β-oxidation by infusion 1 mM octanoate, a medium chain length fatty acid exclusively β-oxidized within the mitochondrial matrix, we observed strong and essentially linear relationships between work output and 14CO2/H14CO− 3 production from [1-14C]pyr-uvate, but only at supraphysiological pyruvate concentrations around 1 mM (60). These PDH flux-work relations occurred despite the fact that 1 mM octanoate is a mild uncoupler in the heart. In the isolated working guinea pig heart the uncoupling effect of 1 mM octanoate was evidenced by lowered myocardial energy state (87), a 67% increase in oxygen uptake unrelated to ventricular work output or inotropy, up to 80% decrease in external work efficiency (62), 32% depletion of myocardial ATP stores and a 109% increase in intracellular Pi levels. In non-working Langendorff rat hearts millimolar octanoate reduced the P/O ratio indicating partial uncoupling of oxidative phosphorylation (87). Despite these unfavorable energetic conditions cardiac 14CO2/H14CO− 3 production from [1-14C]pyruvate remained substantially dependent on the arterial pyruvate concentration (0.05 to 1.0 mM) and on the ventricular inotropic state induced by 0.08 μM NE as well (60). As for myocardial β-oxidation from 1 mM [1-14C]octanoate itself, the basal 14CO2/H14CO− 3 production was ≈1.1 μmol/min·g dry wt in presence of 1 mM unlabeled pyruvate, showing that octanoate oxidation accounted for only ≈26% of measured myocardial oxygen uptake. During 0.08 μM NE infusion octanoate oxidation increased ≈100% and simultaneously was a function of the actual ventricular work output (60, 62). Conversely, the basal rate of pyruvate oxidation under these conditions was ≈9 μmol/min·g dry wt, which accounted for ≈50% of myocardial oxygen uptake, indicating that pyruvate was preferred over equimolar octanoate as energy source under these conditions. However, in contrast to octanoate oxidation, 14CO2/H14CO− 3 production from [1-14C]pyruvate did not appreciably increase during NE stimulation in presence of 1 mM octanoate when rates were corrected for work output; this was presumably due in part to intracellular dilution of [1-14C]pyruvate caused by adrenergic mobilization of glycogen yielding unlabeled glycolytic pyruvate (for further discussion, see next section). Nevertheless, when preload was increased with 1 mM [1-14C]pyruvate, the 14CO2/H14CO− 3 production was highly work dependent over the entire physiological work range both before and during NE stimulation, and this was the case despite the presence of high rates of FFA β-oxidation(60). When the arterial [1-14C]pyruvate concentration was reduced to below physiological levels, i.e. to 0.05 mM, we observed an 8–10-fold decrease in 14CO2/H14CO− 3 production and that 1 mM octanoate greatly weakened, albeit not fully abolished, the stimulatory effects of work output or NE on 14CO2/H14CO− 3 production. On the other hand, when the [1-14C]pyruvate concentration was raised to the physiological level of 0.2 mM, a moderate work dependence of 14CO2/H14CO− 3 production was present. As already mentioned, raising [1-14C]pyruvate level further to 1 mM tightly coupled 14CO2/H14CO− 3 production from [1-14C]pyruvate to work output despite high rates of FFA β-oxidation(60).
In summary, for a given system metabolic state, the rates of cardiac mitochondrial 14CO2/H14CO− 3 production from 1-14C-labeled pyruvate are not only a function of the arterial pyruvate concentration but typically also directly dependent on mechanical ventricular function and inotropic state. This PDH flux-work output relationship manifests itself with at least a minute-by-minute time resolution. The oxidative pyruvate fluxes are driven by the mitochondrial respiration rate in the physiologically performing heart. Under near-physiological conditions the demand-dependence of cardiac oxidative pyruvate flux is significantly modulated, but not abolished even when substantial PDHa inactivation and PDH flux product inhibition are present due to ketonemia, high levels of FFA or millimolar acetate (58). Inotropic agents like catecholamines, calcium or L-type calcium channel blockers acutely and rapidly alter myocardial energy demand to which PDH fluxes adapt with at least a minute-by-minute temporal resolution. In the absence of metabolic downregulation of PDHa or severe product inhibition of PDH flux (simulated by 1 mM pyruvate or 5 mM glucose as sole substrates in presence of insulin), the adaptive responses of the PDH flux to altered work output or inotropy can fully account for changes cardiac mitochon-drial respiration and myocardial oxygen demand.
Steady State Kinetics and Concentration Dependencies of [1-14C]Pyruvate Uptake and 14CO2/ H14CO− 3 Production in the Physiologically Performing Heart.
The remarkable adaptability of cardiac oxidative pyruvate fluxes under greatly different functional and metabolic states was further examined in protocols designed to characterize the steady state kinetics of pyruvate uptake and oxidative pyruvate flux, as well as the effectiveness of pyruvate to induce oxidative pyruvate metabolism in the working heart. This was accomplished by applying Michaelis-Menten type analyses (20, 46, 88) to data from special titration protocols that systematically examined the effects of altered pyruvate concentration on pyruvate uptake and oxidation; these titrations comprised 4–5 steps, starting at about 0.1 mM and ending at 4–5 mM, each step lasting 8 min to achieve metabolic steady states and isotope equilibration. The specific activity of arterial [1-14C]pyruvate was held constant when the pyruvate concentrations were changed. The observed steady state concentration- 14CO2/H14CO− 3 rate relations permitted (legend to Table 2) estimations of the half-maximum effective pyruvate concentration (S0.5), the theoretical maximum rate (Vmax) and the ratio of Vmax/S0.5 or the slope at S0.5 as indices of the effectiveness of the pyruvate “agonist” to induce metabolic 14CO2/H14CO− 3 production. To minimize the effect of work output during the titrations, the hearts were perfused at constant preload (left atrial filling pressure 10–12 cmH2O) and afterload (aortic pressure 85–90 cmH2O) in the absence or presence of a constant NE concentration as indicated in Table 2.
Table 2 summarizes the results by listing the non-linear estimates for S0.5, Vmax, Vmax/S0.5 ratio and slope at S0.5 under the six system metabolic states already mentioned: Hypoglycemia (rows 1–3), Normoglycemia (rows 4, 5), normal lactate levels (row 6), high lactate levels (rows 7, 8), normal β-oxidation of free fatty acids (FFA, 0.2 mM octanoate; rows 9, 10) and ketonemia (rows 11, 12). Each metabolic condition (Table 2, columns 1, 2) required its own specific control (controls 1–4). In the pyruvate uptake studies, arterial pyruvate induced a saturable concentration-dependent net uptake; to examine the possible role of transport competition between pyruvate and lactate at the MCT, a proton-symport system on both the cell membrane and the inner mitochondrial membrane (80, 82, 84, 89), pyruvate titrations were also done in the absence and presence of 1, 5 and 10 mM L-lactate (81, 84, 90). These experiments were performed to re-examine the importance of this pyruvate-lactate transport competition at the molecular level under the conditions of the physiologically working heart, with and without altered inotropic states (20).
Normoglycemia.
In the normoglycemic “resting” working hearts (Table 2, control 2, row 5), the estimated S0.5 for steady state pyruvate uptake was relatively high, ≈0.7 mM, and was not significantly different from the S0.5 for steady state pyruvate oxidation (14CO2/H14CO− 3 production), ≈0.95 mM, under the same system metabolic condition (Table 2, row 4), indicating that pyruvate oxidation from exogenous pyruvate can readily keep pace with pyruvate uptake under the conditions of simulated normoglycemia. In isolated guinea pig and rat cardiomyo-cytes in culture, the Michaelis-Menten Km for initial pyruvate uptake was much lower, 0.07 mM and 0.2 mM, respectively (91), than the current steady state S0.5 estimates in the working hearts. Note that, in the working hearts, the estimated Vmax for pyruvate uptake matched measured total cardiac PDH activity (see Table 1); however, when the Vmax of 14CO2/H14CO− 3 production from [1-14C]pyruvate was compared to the Vmax of net pyruvate uptake, it became clear that the Vmax for uptake was ≈2-fold larger than Vmax for 14CO2/H14CO− 3 production. This substantially greater uptake relative to oxidation was expected from the intense intracellular pyruvate metabolism unrelated to PDH flux, including anaplerosis by CO2 fixation (54, 78, 79, 92) + pyruvate transamination with alanine + pyruvate reduction to lactate (79, 83) + other less powerful cytosolic pyruvate pathways (46).
Hypoglycemia.
In severe hypoglycemia, simulated by supplying exogenous pyruvate as sole substrate, the S0.5 for 14CO2/H14CO− 3 production from [1-14C]pyruvate decreased substantially from ≈0.95 mM to ≈0.3 mM without affecting Vmax (compare control 2, row 4 with control 1, row 1, Table 2); this reflected an increase in measurable 14CO2/H14CO− 3 production during hypoglycemia, which was most likely due to minimal intracellular dilution of labeled pyruvate originating from the arterial [1-14C]pyruvate in the absence of unlabeled glucose. In the presence of normal levels of unlabeled glucose (control 2, row 4), unlabeled pyruvate is generated via glycolysis from glucose, due to glycogen mobilization (26), and presumably also in the alanine transaminase reaction. It is interesting that the moderate hypoglycemia condition with 1 mM (instead of the physiological 5 mM) glucose (Table 2, row 3) also reduced significantly the S0.5 for 14CO2/H14CO− 3 production from [1-14C]pyruvate when compared to the normo-glycemia control (Table 2, row 4). Moreover, when the glucose-free hearts were stimulated by NE (Table 2, row 2), S0.5 for 14CO2/H14CO− 3 production increased > 3-fold into the range of the S0.5 for the normoglycemic hearts (Table 2, row 4); this could be explained by the fact that NE mobilized unlabeled glycogen, which generated unlabeled cytosolic pyruvate via glycolysis, thus diluting intracellularly the label taken up from arterial [1-14C]pyruvate. Increased proteolysis during NE infusion also could have generated unlabeled alanine, which is in near-equilibrium with pyruvate via cytosolic transaminases. In this context the specific activity of alanine has been proposed as a more useful index of the specific activity of oxidized pyruvate than the specific activity of L-lactate in hearts that are perfused with exogenous pyruvate (26), a condition similar to blood perfusion in vivo where plasma pyruvate levels normally are ≈0.08–0.25 mM (78).
High Lactate Levels.
In simulated lactatemia (Table 2, rows 6–8), the presence of normal (1 mM) and high (5–10 mM) L-lactate in the arterial perfusion medium did not decrease Vmax of pyruvate uptake and also did not increase the estimated S0.5. While this was unexpected based on the known transport competition kinetics, the Vmax/S0.5 ratio and the slope at S0.5 for the working hearts decreased significantly and steadily as a function of the arterial L-lactate level, demonstrating that the effectiveness of exogenous pyruvate to stimulate pyruvate net uptake was decreasing with increasing L-lactate levels. Since Vmax did not decrease under these conditions, the results are consistent with competitive transport inhibition at the sarcolemmal MCT (82) under the conditions of the working heart. On the other hand, the fact that S0.5 did not increase with millimolar arterial L-lactate suggests that guinea pig cardiomyocytes (as well as rat cardiomyocytes) have a high-affinity pyruvate uptake mechanism, but a relatively low-affinity lactate uptake system (91; unpublished data). In other words, L-lactate is a weak inhibitor of cardiac pyruvate uptake, while pyruvate is a strong inhibitor of cardiac lactate uptake in both isolated myocytes as well as the working heart.
β-Oxidation.
What is the effect of physiological FFA β-oxidation on the steady state kinetics of cardiac oxidative pyruvate fluxes in the physiologically working hearts? We simulated normal β-oxidation of FFA by infusing sub-millimolar 0.2 mM octanoate, not the uncoupling millimolar concentrations of octanoate (normal FFA blood level is ≈0.6 mM (54)). We observed unexpectedly that the estimated Vmax for pyruvate oxidation was 37% higher, not lower (Table 2, row 9), than the Vmax of the “resting” normoglycemic control (P < 0.01 relative to Table 2, control 2, row 4). On the other hand, in the octanoate hearts, the observed S0.5 of ≈0.3 mM was not different from that obtained with pyruvate as sole substrate (Table 2, control 1, row 1), which suggested unrestricted and “undiluted” pyruvate uptake plus 14CO2/H14CO− 3 production from [1-14C]pyruvate during normal FFA oxidation in the working heart. The observed increase in Vmax for 14CO2/ H14CO− 3 production from [1-14C]pyruvate at physiologically low S0.5 reflected stimulation of cardiac pyruvate oxidation due to low levels of β-oxidation. Since we used submillimolar 0.2 mM octanoate (not uncoupling octanoate), our finding can be reconciled with reports from other laboratories showing, in the porcine heart, that 1 mM octanoate strongly inhibited, not stimulated pyruvate oxidation (54) and, in the non-working rat Langendorff heart, that 2 mM octanoate completely abolished 13CO2 production from 2 mM [1-13C]pyruvate (93). Similarly, our finding of enhanced pyruvate oxidation during low level β-oxidation is also consistent with data from perfused rat liver and isolated rat hepatocytes, which demonstrated stimulation, not inhibition of pyruvate oxidation by submillimolar octanoate or palmitate. Investigators of these hepatic protocols suggested that the mechanism of the FFA-enhancement of pyruvate oxidation is a stimulation of mitochondrial pyruvate uptake (import) in exchange for mitochondrially generated acetoacetate export (76, 92). When the low-level β-oxidizing guinea pig hearts were stressed by near maximally effective NE (Table 2, row 10), the estimated Vmax of pyruvate oxidation equaled both estimated total maximal pyruvate uptake during normoglycemia (Table 2, row 5) and total myocardial PDH activity (see Table 1). This result indicates that during high cardiac work states due to catecholamines, pyruvate uptake is increasingly partitioned in favor of mitochondrial pyruvate oxidation, presumably at the expense of pyruvate carboxylation (anaplerosis) and other non-PDH related pathways. Such metabolic re-focusing is advantageous energetically and also consistent with other metabolic effects of cardiac NE stimulation, e.g., the significant increase intracellular pyruvate levels from ≈60 μM to ≈125 μM in normoglycemia as well as during β-oxidation (26). Such intracellular pyruvate concentration changes may well be cell-physiologically relevant, since they overlap with the Ki for pyruvate inhibition of PDH kinase (≈30 μM) in porcine cardiac PDH (56) and also with the 90 μM extramitochondrial pyruvate that can activate PDH by 37% in isolated rat liver mitochondria (25). Also consistent with such NE-dependent intracellular pyruvate accumulations are our findings in the working guinea pig hearts that show that cardiac pyruvate decarboxylation can fully meet the myocardial energy demand during maximum adrenergic stimulation in simulated normoglycemia or during low-level β-oxidation, i.e. when intracellular pyruvate levels are in the low physiological range of 60–120 μM in normoglycemic hearts or somewhat higher, ≈155–250 μM, in glucose + 0.2–0.3 mM pyruvate- or glucose+ 0.5 mM octanoate perfused working hearts (20, 58, 60).
Ketonemia.
Finally, the observed steady state kinetics data from the ketonemic hearts (Table 2, rows 11, 12) were distinctly different from all other system metabolic conditions reported on in Table 2. Most importantly perhaps, in the absence of NE, simulated ketonemia raised the S0.5 for 14CO2/H14CO− 3 production from [1-14C]pyruvate 14-fold relative to the pyruvate-alone condition (Table 2, control 1) or about 5-fold relative to the normoglycemia condition (Table 2, control 2). Simultaneously, both the Vmax/S0.5 ratio and the slope at S0.5 decreased almost 95% (compare row 11 with control 1 of Table 2), indicating a greatly reduced effectiveness of arterial pyruvate to induce net pyruvate oxidation during ketonemia. This high degree of inhibition was consistent with the very low absolute rates of 14CO2/H14CO− 3 production from [1-14C]pyruvate in those ketonemic hearts that metabolized pyruvate at low physiological levels (Fig. 10, panels B, C). However, there was no decrease in estimated Vmax in the ketonemic control hearts (Table 2, control 4) when compared to the non-ketonemic controls with glucose or pyruvate (Table 2, controls 1 or 2). Such kinetics suggested that a major limitation for pyruvate oxidation during severe ketonemia was impairment of uptake (transport), possibly at both the sarcolemmal and mitochondrial MCTs, not a near-100% inactivation of PDH (see Table 1) and/or complete product inhibition of PDH flux by accumulated mitochondrial NADH and acetyl CoA. Of special interest was that during NE stimulation the kinetics of pyruvate oxidation improved dramatically in the ketonemic hearts (Table 2, row 12). Figures 10B, C had already revealed that the NE-mediated increases in 14CO2/H14CO− 3 production from [1-14C]pyruvate during ketonemia were much larger than the associated increases in work output and myocardial oxygen uptake. Such enhanced stimulatory NE-effect is reflected in the observed 3-fold decrease of S0.5 coupled with 5-fold increases in both the Vmax/S0.5 ratio and slope at S0.5 (Table 2, rows 11, 12). This kinetics is consistent with Table 1 showing that during NE stimulation 14CO2/H14CO− 3 production from [1-14C]pyruvate not only increased in absolute terms but also relative to PDHa activity and oxygen uptake, respectively.
One explanation for the remarkable enhancement by NE of pyruvate oxidation during severe ketonemia is the fact that cardiac D-β-hydroxybutyrate oxidation remained nearly constant (62) instead of responding with demand-dependent increase during adrenergic stimulation (63, 64). Taegtmeyer reported that cardiac ketone body oxidation alone failed to support hemodynamic and metabolic stability of working rat hearts (94) and we observed (unpublished) that D,L-β-hydroxybutyrate is a poor substrate compared to pyruvate, L-lactate or submillimolar octanoate for restoration of cardiac work output in the guinea pig heart after prior hemodynamic failure due to energetic depletion. Another mechanism for the remarkable NE stimulation of cardiac pyruvate oxidation during ketonemia is likely related to increased mitochondrial respiration due to NE, which is plausible, since increased mitochondrial respiration increases the proton gradient across the inner mitochondrial membrane (alkaline inside) (95), thereby enhancing proton-compensated pyruvate import via the MCT into the mitochondrial matrix. In other studies we estimated the capacity of the cardiac mitochondrial pyruvate carrier in situ and found that it is ≈2.5–3-fold higher than total cardiac PDH activity; this suggested that the MCT-mediated mitochondrial pyruvate influx (84) is not very effective in limiting pyruvate oxidation in the intact heart, at least not in the absence of ketonemia nor during normal FFA β-oxidation (20). With respect to pyruvate transport from the extracellular space into the cytosolic compartment, it is doubtful that NE stimulated the pyruvate transport across the sarcolemma into the cytosol; this is the case because adrenergic stimulation slightly acidifies, not alkalizes, the cytosol thus slightly decreasing, not increasing, the driving force for MCT-mediated proton-compensated pyruvate import across the cell membrane (62). Regardless of the exact mechanism(s) that effect the disproportionately strong stimulation of PDH flux during physiological adrenergic stimulations of ketonemic hearts, our data show that NE-induced increases in myocardial energy demand can, to a considerable extent, override the metabolic regulation of the PDH system by ketone bodies and other competing energy substrates.
Implications for Cardiac Metabolic Imaging Based on the Use of Hyperpolarized [1-13C]Pyruvate Tracer
Alanine Labeling Is a Better Indicator than Lactate for Mitochondrially Oxidized Pyruvate.
The cardiac pyruvate literature and our current data also provide pragmatic information for the optimization of the hyper-polarized [1-13C]pyruvate technology in metabolic imaging of the heart in vivo in future. It is has been demonstrated that substantial output of carbon-labeled CO2/HCO− 3 from exogenous 13C-labeled pyruvate, even when applied in low physiological concentrations, as well as substantial parallel labelings of cytosolic lactate or alanine can be obtained within 5 min after the start of the exogenous pyruvate pulse, although full isotope equilibrations require at least 8–12 min in rat (Fig. 11) and guinea pig myocardium (26, 79) (in canine myocardium even more time is required (83)). Although the [1-14C]pyruvate pulse will likely label the intracellular lactate pool in synchrony with14CO2/H14CO− 3 output in the working heart (26), the measured steady state specific activity of lactate proved to substantially underestimate the specific activity of mitochondrially oxidized pyruvate, i.e. by a factor of 2–4 (62, 79). On the other hand, the specific activity of myocardial alanine after reaching the labeling plateau at about 5 min reflected ≈70% of that of mitochondrially oxidized pyruvate in the rat heart (79). A several-fold difference between lactate and alanine labeling using [3-13C]pyruvate as precursor was confirmed in the dog heart in vivo (83). Also in perfused rat liver, carbon tracer studies detected “unconventional” labeling of pyruvate and lactate as reported by Müllhofer and colleagues (73, 75). In perfused guinea pig and rat hearts the 5 min time frame for near-fully labeling alanine overlapped with the 4 min time span required for 14CO2/H14CO− 3 production from [1-14C]pyruvate to reach ≈95% of steady state output (26, 78). Taken together these temporal correlations in labeling patterns using exogenous physiological pyruvate as precursor support the conclusion that lactate and pyruvate are subcompartmented in the cytoplasm of the myocardium; this becomes particularly evident in the presence of exogenous pyruvate (26, 78) but seems negligible in the unphysiological condition of glucose (or glycogen) being the sole or main source for intracellular pyruvate (26).
To account for this type of physiological pyruvate subcompartmentation in assessing the in vivo heart PDH flux (where exogenous pyruvate is always present), it seems reasonable to refer the [1-13C]pyruvate pulse-dependent 13CO2/H13CO− 3 signal to the simultaneously obtained [13C]alanine signal. We believe such correction would provide the best rationale for the use of the 13carbon-labeled CO2/HCO− 3 from [1-13C]pyruvate as an index of PDH flux. In summary, the intensity of the recordable 13CO2/H13CO− 3 signal during a hyperpolarized [1-13C]pyruvate pulse labeling can be expected to be modulated significantly by variables such as the arterial pyruvate concentration, the achieved specific activity of mitochondrially oxidized [1-13C]pyruvate, the presence or absence of competing system metabolic substrates (ketone bodies, FFA, L-lactate, acetate) and finally also the instantaneous work and inotropic states of the myocardium. Under pathological conditions such as ischemia, hypoxia and reperfusion, an additional variable will be the generation of unlabeled pyruvate from glycogen mobilization, and in severe cases possibly also from proteolysis involving alanine trans-aminations.
Implications for Studies of PDH-Flux In Vivo.
How can we optimize the signal to noise ratio and the interpretability of the 13CO2/H13CO− 3 signal under the conditions of the in-vivo heart challenged by a hyper-polarized [1-13C]pyruvate pulse? The steady state kinetics of Table 2, the transitory and steady state correlations in Figure 10 and the known partitioning of cardiac pyruvate metabolism suggest the following considerations: i) minimize pyruvate transport competition by ketone bodies or related compounds, ii) relieve downregulation of PDH activity and PDH flux product inhibition by NADH and acetyl-CoA, iii) minimize intracellular dilution of the exogenous [1-13C]pyruvate tracer by minimizing cardiac glycogen mobilization and the rate of glycolysis, iv) expose the myocardium for at least 4–5 min with the tracer before recording the 13C-NMR signals from 13CO2/H13CO− 3 and [13C]alanine, and v) raise the tracer [1-13C]pyruvate concentration such that the hyperpolarized pyruvate pulse establishes an intracoronary pyruvate concentration well above the S0.5 for any given system metabolic state (c.f. Table 2).
HD-MRI for Cardiac Patients?
In practice, such conditions could be approximated by not allowing the patient to fast overnight, by reducing emotional and psychological stress (lowering blood catecholamines that mobilize cardiac glycogen and stimulate glycolysis), and perhaps by pharmacologically activating the PDH by dichloroacetate prior to the pulse labeling. To minimize the extracellular dilution of the injected tracer by the pre-existing blood pyruvate it would be useful to catheterize the coronary artery using cardiological techniques; if this is not practical, the next best catheter placement would be intraventricularly in the left ventricle, which is clinical practice in contrast media applications. In the data analysis following the pulse labeling with [1-13C]pyruvate, the 13CO2/H13CO− 3 and [13C]alanine myocardial signals should be treated semi-quantitatively by densitometry and by evaluating the ratio of (13CO2+H13CO− 3)/[13C]alanine, which appears to be the currently best estimate of PDH flux under the conditions existing at the time of the pulse labeling by [1-13C]pyruvate. Some form of correction for intracellular dilution of the pyruvate tracer is even more important when patients present with cardiac ischemia or hypoxia. These pathological conditions induce acute cardiac glycogen mobilization or, if they are chronic, also stimulate proteolysis; both glycogen breakdown and proteolysis use metabolic pathways that generate unlabeled pyruvate and alanine thus diluting intracellular [1-13C]pyruvate and thereby reducing the recordable 13CO2/H13CO− 3. The analysis should ideally be done using data obtained only after the [1-13C]pyruvate pulse had been in contact with the heart for at least 4–5 min, and with a concentration near or better above the S0.5 of the prevailing system metabolic condition of the patient (examples for various S0.5 values for different system metabolic states are presented in Table 2).
“Hyperpolarized” Cardiology.
Once the hyperpolarized [1-13C]pyruvate technology is perfected for clinical cardiological applications, it likely will provide the information to rationally evaluate and reliably interpret the 13CO2/H13CO− 3 signal from [1-13C]pyruvate, a key index of the oxidative function of the cardiac mitochondria in situ. It may be mentioned in this context that i.v. or i.a. pyruvate up to low millimolar concentrations (1–3 mM) has no known toxicities in healthy volunteers, cardiac patients and swine, at least not in short-term applications up to 2 hr; thus the hyperpolarized [1-13C]pyruvate technology appears to promise minimal or no toxicity in the clinical setting (96–101).
Conclusions
By enhancing MR signal 10,000 to 100,000 times and demonstrating feasibility for cardiac imaging of metabolic events, both PASADENA and DNP tracers present themselves as the MRI and MRS modalities of the future, with a new immediacy for early diagnosis of cardiac disease states.
We propose the more general term High Definition MRI (HD-MRI) be used to describe these and other modalities described in this review. By closing the SNR gap, HD-MRI applied to molecular imaging will allow MR to compete favorably with PET and CT.
HD-MRI applied to clinical cardiology—by any or all of the techniques presented in Figure 2—may be closer than we think.
For optimization of an in vivo real-time metabolic image of the PDH system in the working heart, using the 1-13C-labeled pyruvate after hyperpolarization, the metabolic system status should be known since it can greatly affect the kinetics and magnitude of the recordable cardiac 13CO2/H13CO− 3 signal.
Because of extracellular and intracellular dilutions of the applied 1-13C-labeled pyruvate bolus in vivo, currently the best estimate of oxidized mitochondrial pyruvate (indicating PDH flux) appears to come from the 13CO2/H13CO− 3 signal related to the simultaneously recorded 13C-alanine signal.
Based on time course studies in the controlled settings of perfused rat and guinea pig hearts utilizing physiological pyruvate at 0.2–0.3 mM, a near-isotope equilibration between the hyperpolarized pyruvate and the cardiac 13CO2/ H13CO− 3 and 13C-alanine signals can probably be reached within 4 to 5 min after start of [1-13C]pyruvate infusion.
HD-MRI with DNP and 1-13C pyruvate appear poised for cardiac metabolic imaging.
Ventricular Work Determines Cardiac Oxidative Pyruvate Fluxes a
Experimental Evidence that Uptake and Oxidation of Pyruvate Are Strongly Dependent on Pyruvate Concentration a
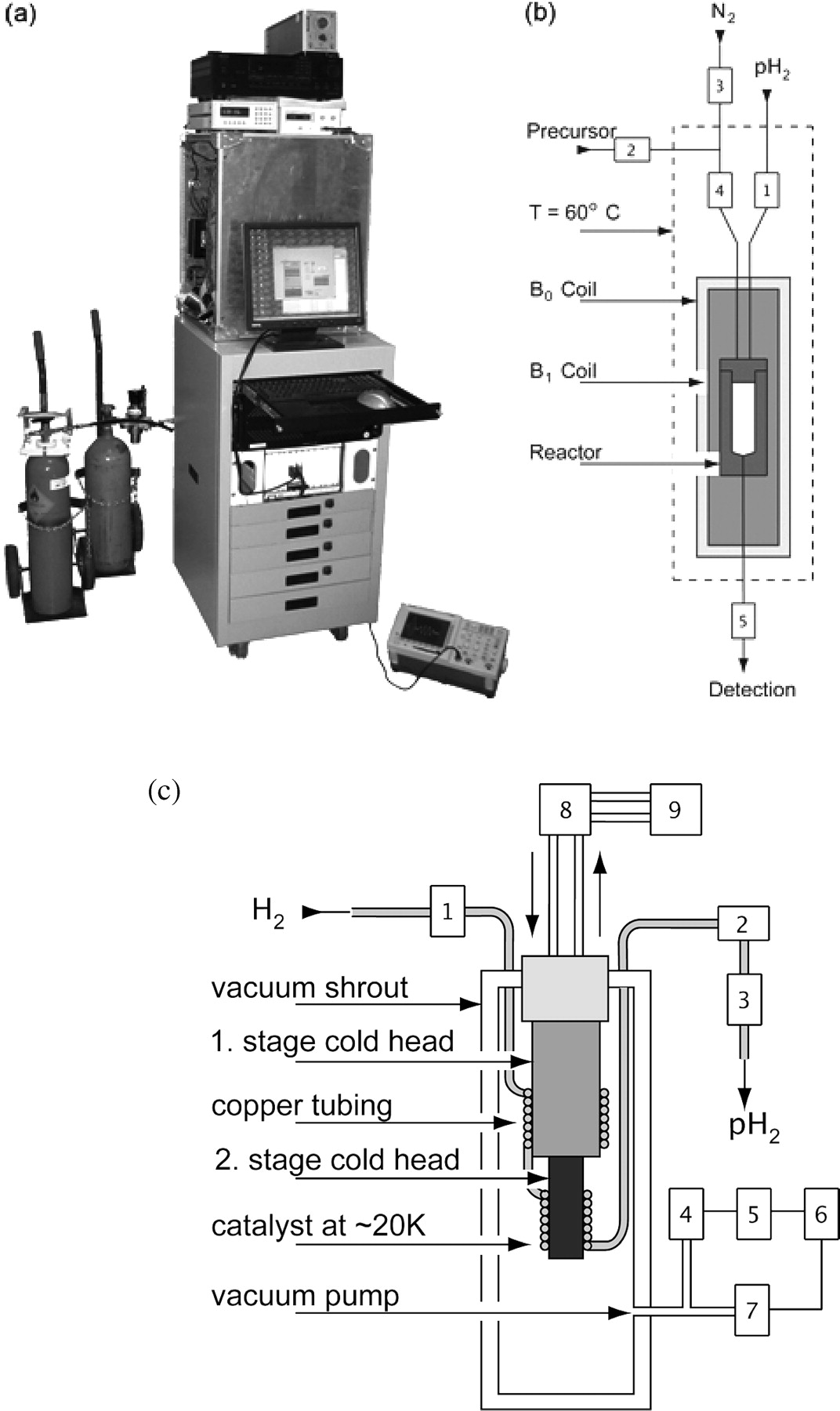
a) Photograph and (b & c) schematic view of the experimental setup. (a) Low-pass filter, r.f. amplifier, gauss meter, Bo power supply and reaction compartment, on top of the rack for the computer and synthesizer, parahydrogen (left) and nitrogen (right) gas tanks (top to bottom). (b) Functional elements in the reaction compartment: B0 coil, B1 coil and reactor. The valves for fluid control are indicated with number {V1}–{V5}. During a PASADENA experiment, (1) the reactor is filled with parahydrogen to a pressure of 10 bar, (2) the precursor molecule is injected through the center of the injection cap and (3) the r.f. spin order transfer sequence is applied after a reaction time of 3 s. Thereafter, the hyperpolarized agent is withdrawn through the end cap and delivered to the detecting MR unit. (c) Schematic of a parahydrogen unit for the PASADENA polarizer. Commercially available hydrogen gas (25% parahydrogen, 75% orthohydrogen) is cooled to ~20 K and passed through a catalyst (granular ferric oxide, A) for > 97% conversion to parahydrogen. The two stages of the cold head are surrounded by copper tubing packed with the catalyst: (1) Pressure regulator for incoming hydrogen; (2) flow meter; (3) flow controller; (4) pressure gauge; (5) pressure display; (6) relay; (7) vacuum pump valve; (8) helium compressor; (9) water cooler.
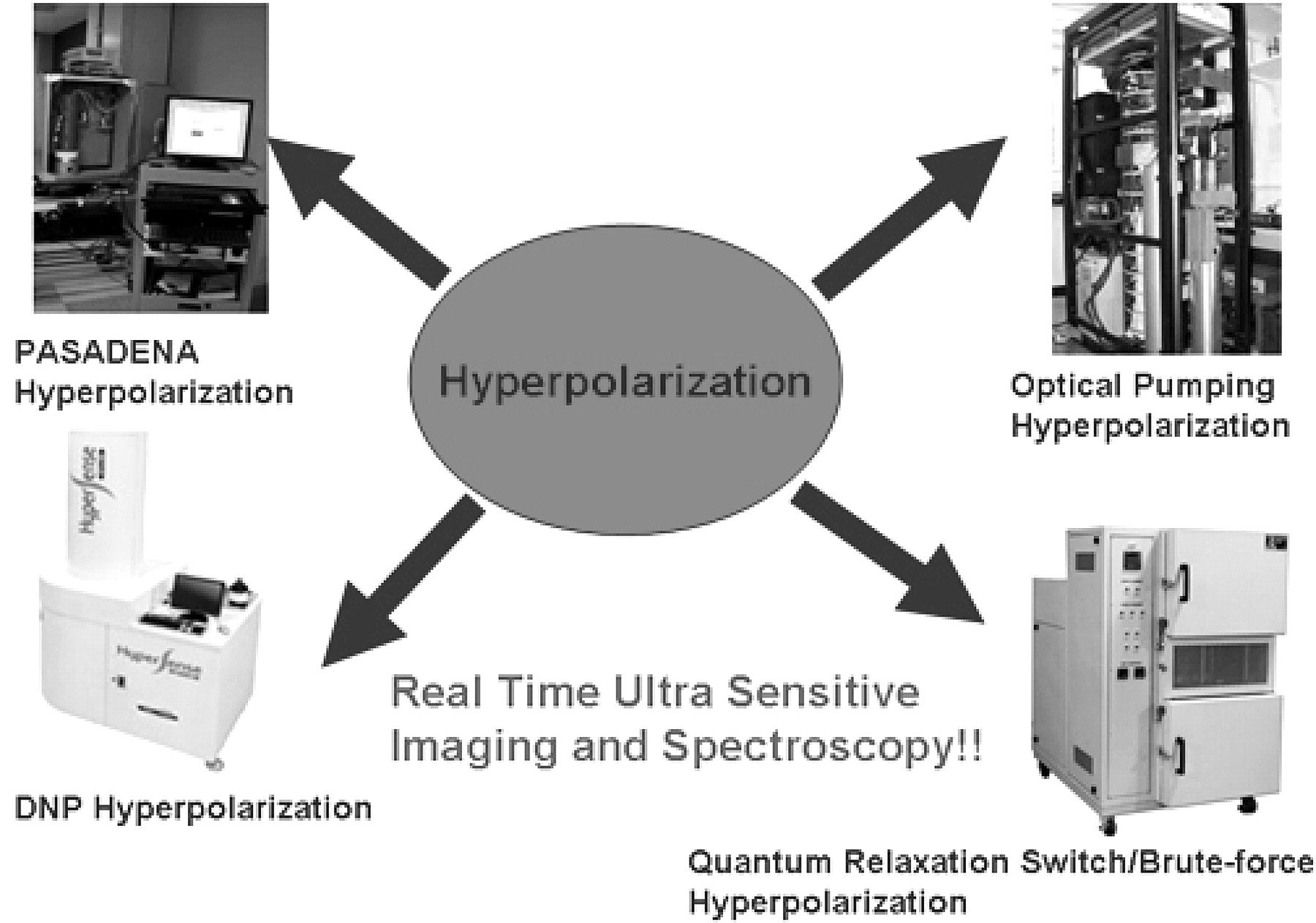
Modalities of hyperpolarization.
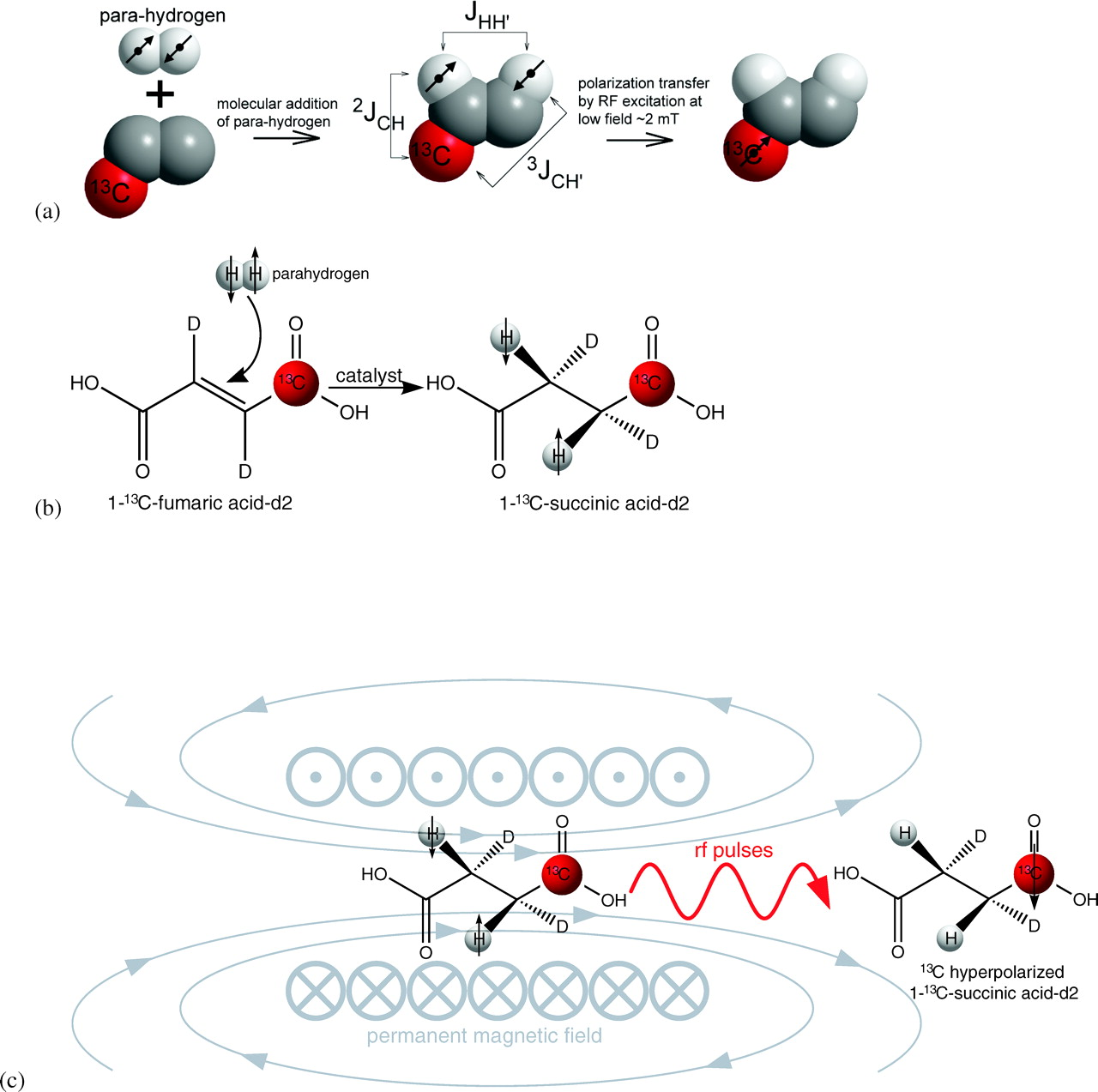
(a) The transfer of spin order from proton of parahydrogen to a 13C nucleus of a biomolecule by r.f. excitation. (b) A specific example of molecular addition of parahydrogen to 1-13C-fumaric acid-d2 to produce 1-13C-succinic acid-d2. (c) The scheme of the spin order transfer from singlet states of parahydrogen spins to 13C1. R.f. pulses are applied to transfer the spin order inside a 1.8 mT electromagnet following by ejection of hyperpolarized 1-13C-succinic acid-d2. The entire process is automated in the polarizer shown in Figure 1.
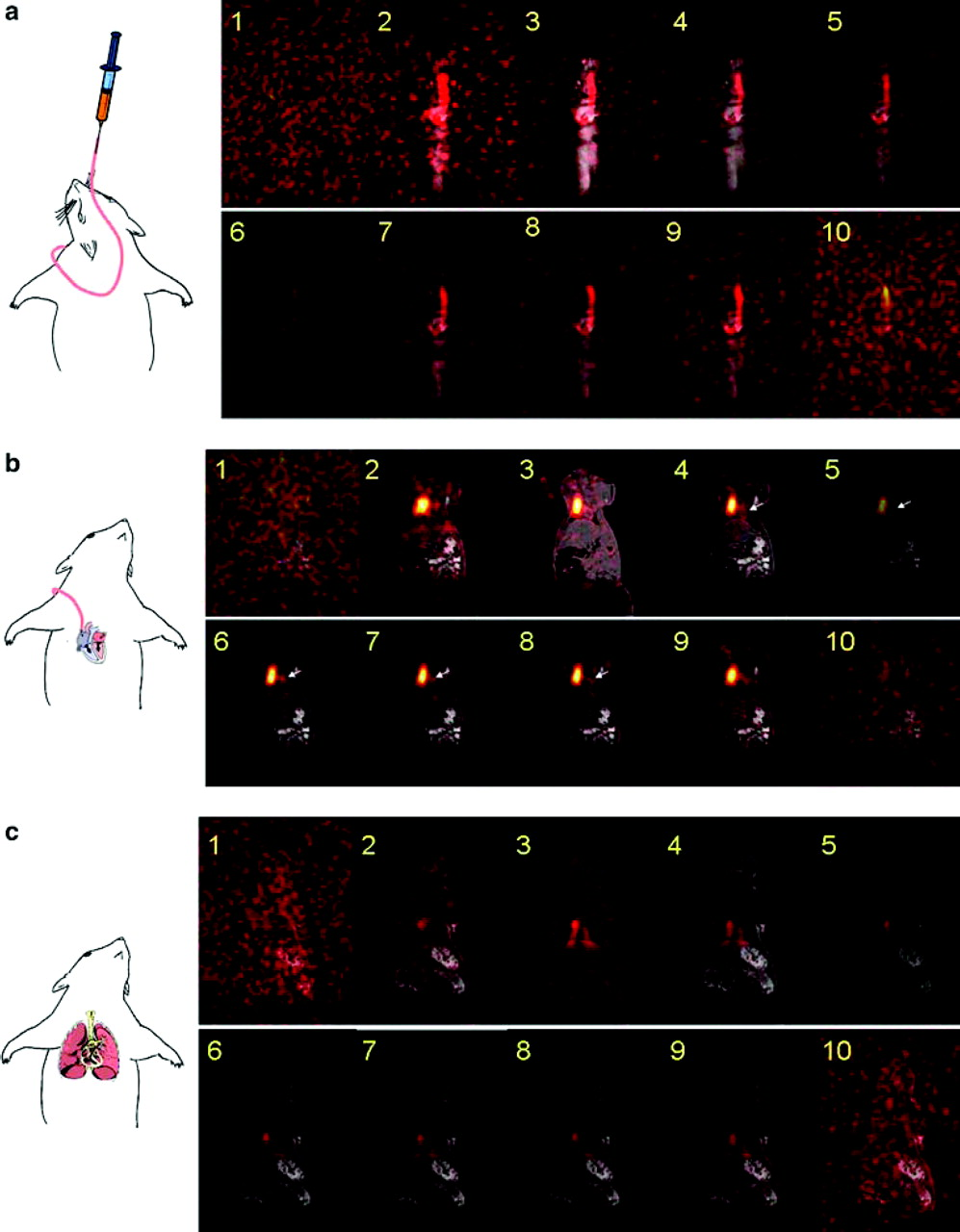
Dynamic hyperpolarized 13C PASADENA MRI in vivo. (a) Each 2D image is selected from the same slice acquired in the first ten acquisitions of 3D imaging. The slice was selected to best visualize the catheter. Each image was acquired in 3.66 seconds; images 1–10 represent the first minute post-infusion of 13C hydroxyethylpropionate (HEP). (b) Time course of arrival of hyperpolarized 13C hydroxyethylpropionate in the heart. (c) Time course of arrival of 13C hydroxyethylpropionate (HEP) in the lungs. Adapted from Bhattacharya P et al., 2005 (14).
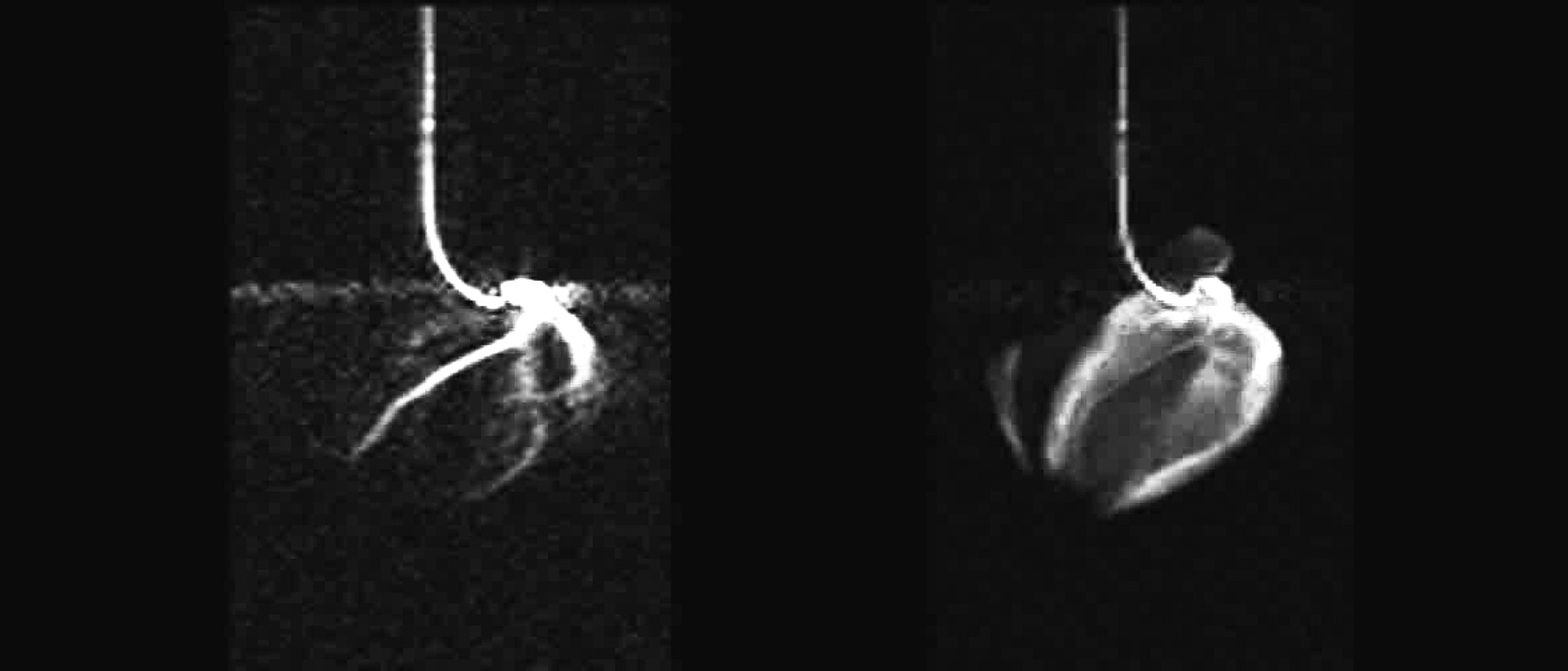
13C TrueFISP imaging of a pig heart on injection of hyperpolarized HEP via a catheter in the coronary artery. Shown (a) are the three coronary arteries left, right and circumflex, and (b) real time myocardial perfusion. Adapted from Mansson S et al., 2006 (16).
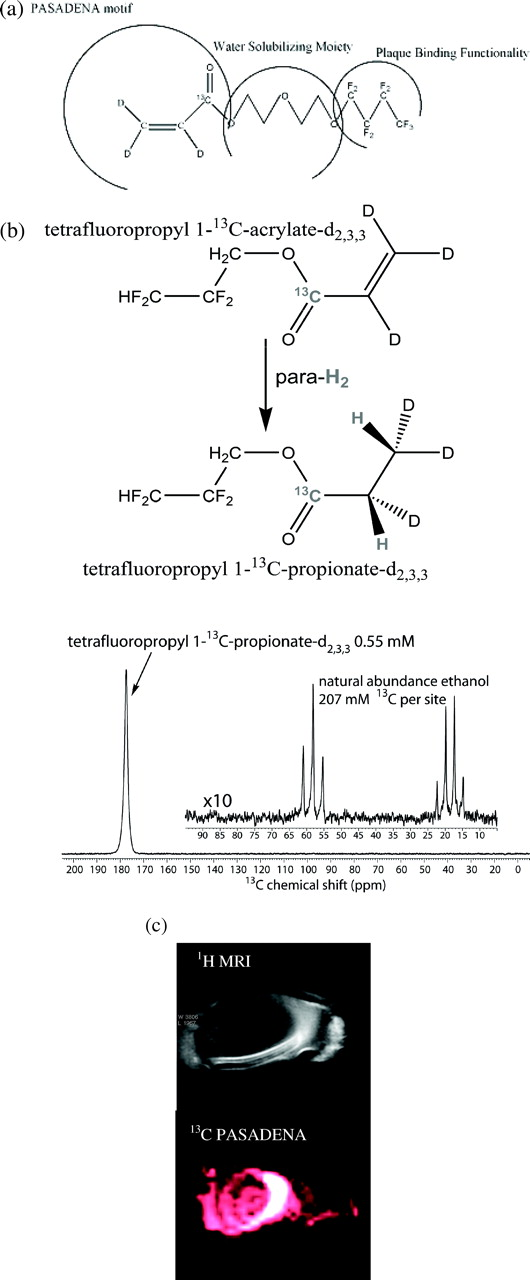
a: General molecular design of a plaque targeting agent. b: Typical 13C hyperpolarized spectrum (P: 25%) of 0.55 mM 1-13C-TFPP-d2,3,3 at 4.7T in vitro. Natural abundance ethanol with 207 mM of 13C per carbon site is used as reference. c: 1H conventional MRI (top) and 13C PASADENA subsecond FIESTA image (bottom) acquired utilizing 1-13C-TFPP-d2,3,3 of an isolated pig aorta acquired at 4.7 T Bruker animal scanner.
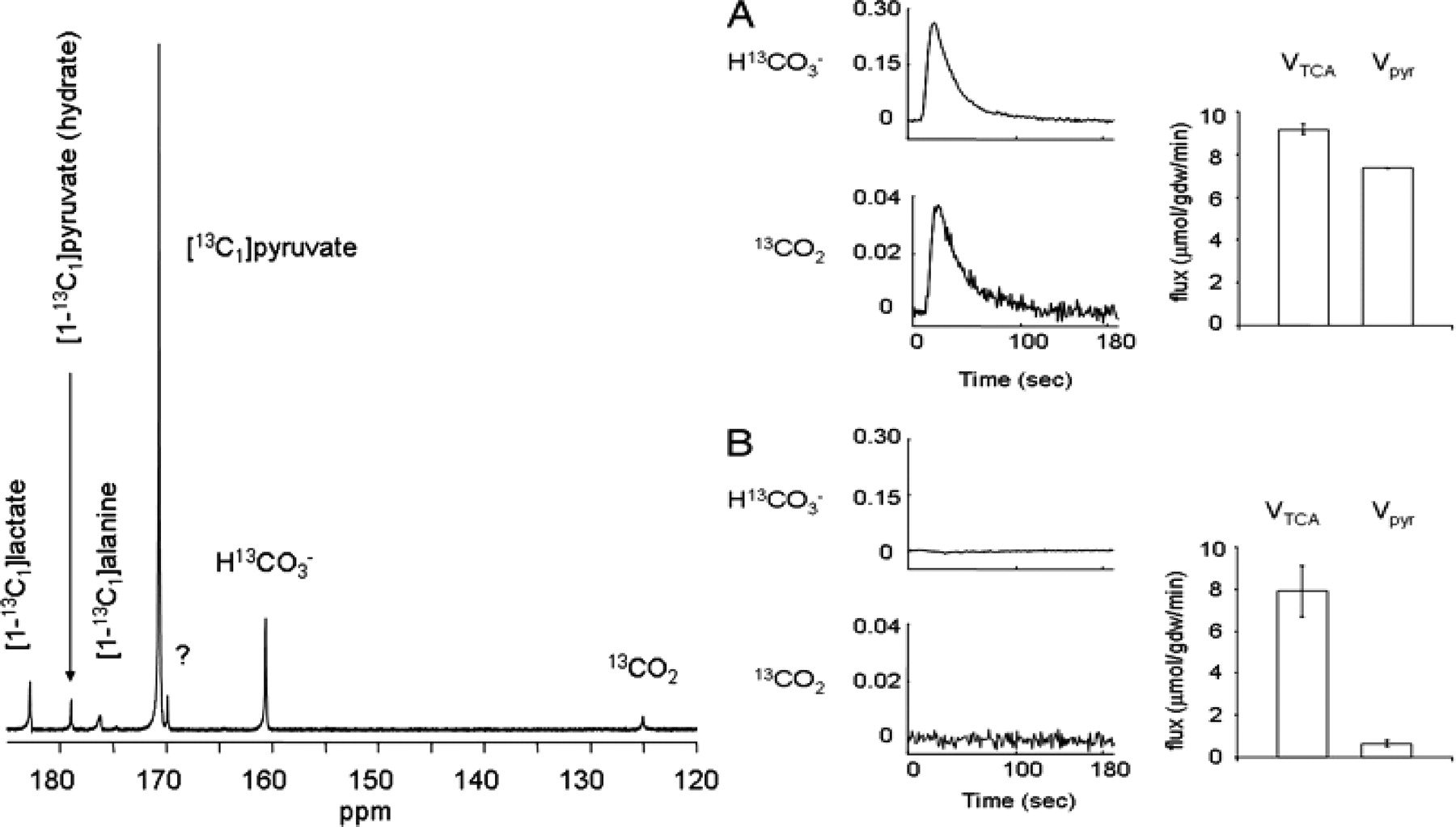
Left: 13C NMR spectrum of an isolated rat heart. This spectrum is the sum of 100 scans. Right: Effect of 2 mM octanoate on appearance of 13CO2 and H13CO3 production from 2 mM [1-13C]pyruvate. The average 13C NMR signals from dissolved 13CO2 and H13CO3 are shown relative to the maximum pyruvate signal from hearts supplied with 2 mM [1-13C1]pyruvate (A) or 2 mM [1-13C1]pyruvate plus 2 mM octanoate and 2 mM propionate (B). Vpyr indicates flux through PDH measured separately with non-hyperpolarized [1-13C]pyruvate in panels A and B. Adapted from Merritt ME et al., 2007 (93).
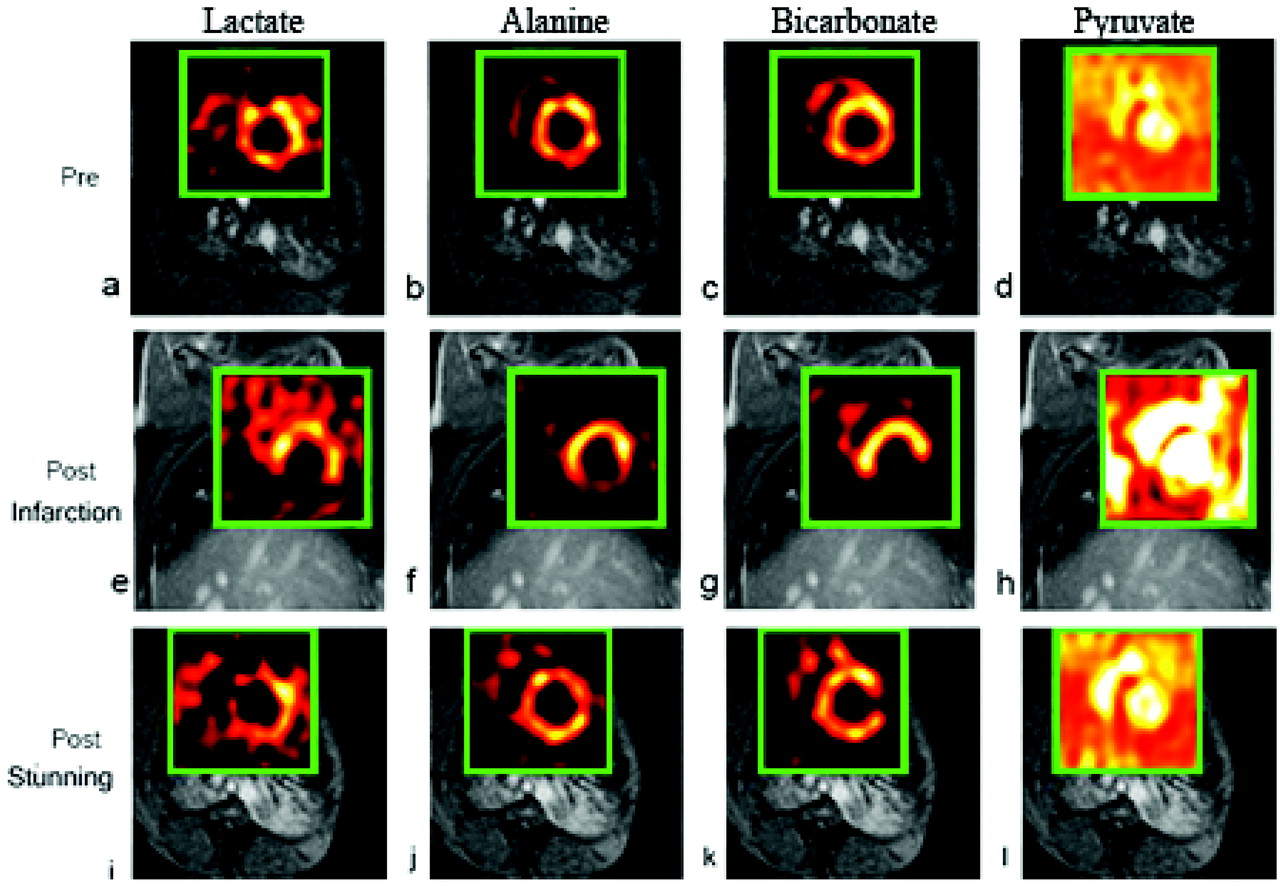
These images illustrate the diagnostic potential of cardiac hyperpolarized [1-13C]pyruvate imaging by NMR detection mediated by the mitochondrial pyruvate dehydrogenase (PDH), the cytosolic lactate dehydrogenase and the alanine transaminases/glutamate-pyruvate transaminase in stunned vs. infarcted myocardium. Mitochondrial respiration (myocardial oxygen uptake) is the ultimate driving force for conversion of 13C-labeled pyruvate into carbon-labeled CO2/HCO− 3 in the PDH reaction (PDH flux = oxidative decarboxylation of pyruvate). The PDH reaction decarboxylates pyruvate generating intramitochondrial acetyl-CoA for the TCA cycle and NADH for the respiratory chain enabling O2 reduction coupled to oxidative phosphorylation of ADP to ATP in the presence of inorganic phosphate. Normal cardiac contractility and work performance in vivo and in vitro obligatorily require mitochondrial oxidative phosphorylation, which can be fully supported by oxidative pyruvate decarboxylation regardless of whether pyruvate is supplied exogenously or generated cytosolically via glycolysis. Also low exogenous physiological pyruvate levels support oxidative pyruvate decarboxylation, which is directly related to cardiac work output (Fig. 7). Thus, under normal conditions, the presence or absence of PDH flux as well as its intensity (speed of the reaction) are indicative of the presence or absence of mitochondrial respiration coupled to oxidative phosphorylation of ATP in the region of interest selected by the 13C MRI analyses. Adapted from Golman K et al., 2008 (45).
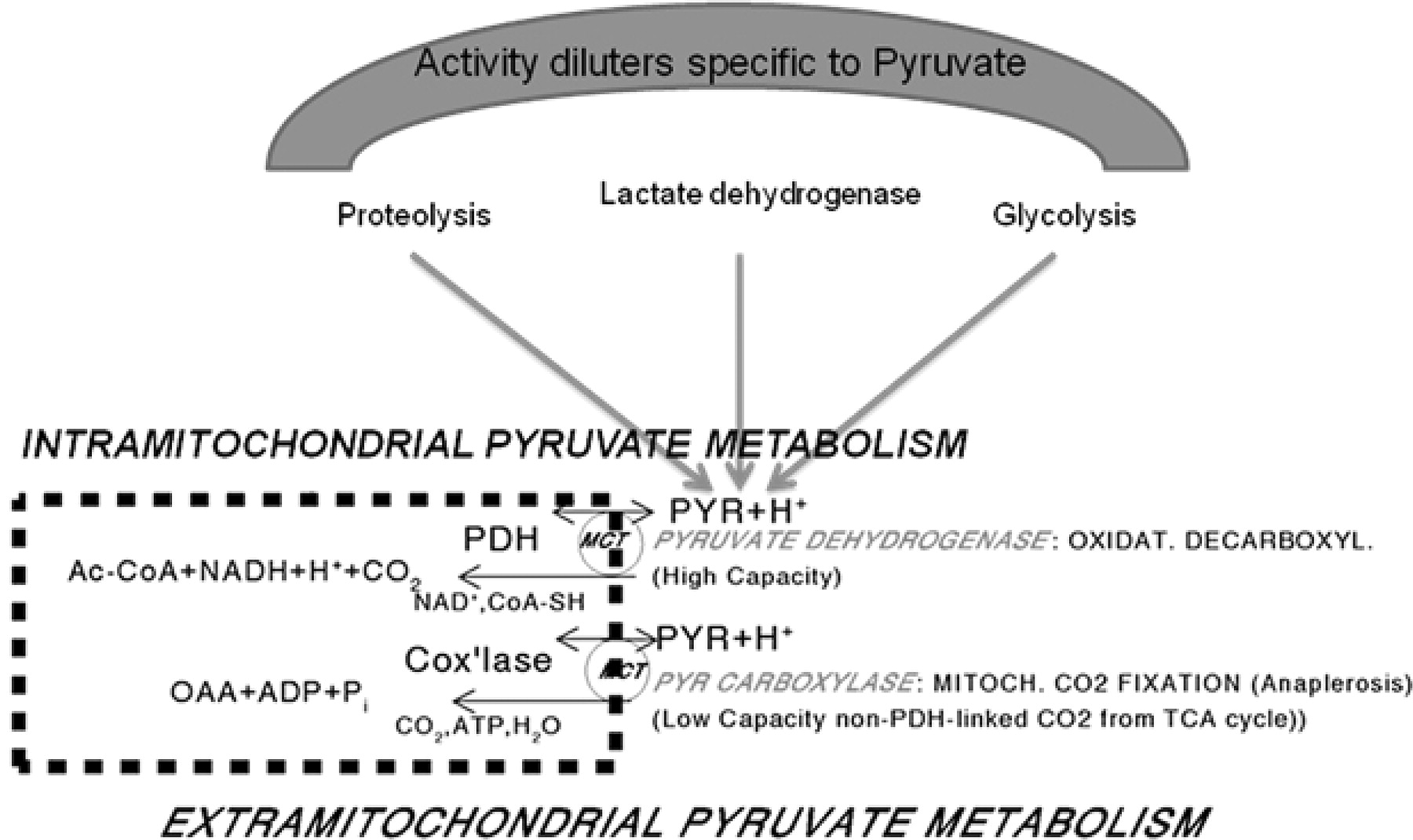
Simplified model of compartmentalized pyruvate metabolism in mammalian cells containing mitochondria. When exogenous hyperpolarized or otherwise labeled pyruvate tracer is applied, its specific activity becomes diluted intracellularly and extramitochondrially by unlabeled pyruvate formed from glycogen and in glycolysis, from blood lactate oxidized to pyruvate via the powerful lactate dehydrogenase and from alanine transamination, especially when net protein catabolism exists. The mitochondrial pyruvate carboxylase system fixes CO2 to labeled pyruvate generating labeled oxaloacetate and citrate intramitochondrially contributing to total labeled CO2/HCO3 formation from pyruvate; this latter rate is not PDH flux and has been estimated to account for about 7–10% of total pyruvate-dependent CO2/HCO3 formation in the working myocardium metabolizing [1-14C]-labeled pyruvate under near-physiological conditions. The known cytoplasmic subcompartmentation of pyruvate and the low-capacity cytosolic malate dehydrogenase (NADPH dependent) are not shown. For further explanations, see text.

Transient and steady state 14CO2 production from physiological levels of exogenous [1-14C]pyruvate by isolated working guinea pig hearts under normoglycemic (5 mM glucose (5 U/L insulin) +0.3 mM pyruvate, A) or ketonemic conditions, B) 2 mM DL-β-hydroxybutyrate +0.3 mM pyruvate, C) 10 mM DL-β-hydroxybutyrate + 0.1 mM pyruvate). Note the large differences in 14CO2 rates between normoglycemic and ketonemic hearts while the tight and direct correlations between instantaneous work output or NE-induced contractility changes and 14CO2 rates are retained also during transition from low to high work states. The data also show that L-lactate release is not tightly correlated with cardiac work or inotropy in the steady states. Metabolic and hemodynamic rates refer to ventricular fresh wet weight.

A study in the in vivo rat heart illustrates the same point; by using hyperpolarized [1-13C]pyruvate as a probe, investigators at Oxford University confirm the downregulation of PDH by fasting, high fat diet and diabetes mellitus (diseased). Adapted from Schroeder MA et al., 2008 (102).
Footnotes
We thank the following for funding: NIH R21CA118509, NCI R01CA122513, NIH R01NS048589, NIH R01HL37067, NIH R01HL29060, NIH R01HL34579, Rudi Schulte Research Institute (RSRI), American Heart Association, American Brain Tumor Association, Tobacco Related Disease Research Program (16KT-44), Gustavus and Louise Pfeiffer Research Foundation, Pasadena Community Foundation, Uniformed Services University of the Health Sciences: RO7638, RO70LO, RO76HB; Intramural Funds of Department of Physiology, Munich; Naval Office of Research and Development, Veterans Administration (G376CD), American Heart Association–Texas Affiliate (92G-155), Bay-Zoltan Foundation for applied research, Sirona Industries for royalties.
Based on a lecture delivered by Dr. Pratip Bhattacharya on Jan 31, 2008 at the invitation of Society of Cardiovascular Magnetic Resonance (SCMR), Beverley Hills, California.
Acknowledgements
We thank Olaf Sommer, David Hartman, Dr. Robert Mallet, Dr. Bernhard Permanetter, Dr. William Perman, Dr. Eduard Chekmenev, Dr. Daniel Weitekamp, Dr. Wanda Reynolds, Dr. Shawn Wagner, Dr. Henry Chan, Thao Tran, Dr. Jan B. Hovener, Valerie Norton, Dr. Damian Tyler, Dr. Alexander Lin and Dr. Keiko Kanamori for helpful discussions. Special thanks to Dr. Heinrich Taegtmeyer for valuable suggestions.