
Abstract
The endothelial glycocalyx (eGlx) constitutes the first barrier to protein in all blood vessels. This is particularly noteworthy in the renal glomerulus, an ultrafiltration barrier. Leakage of protein, such as albumin, across glomerular capillaries results in albumin in the urine (albuminuria). This is a hall mark of kidney disease and can reflect loss of blood vessel integrity in microvascular beds elsewhere. We discuss evidence demonstrating that targeted damage to the glomerular eGlx results in increased glomerular albumin permeability. EGlx is lost in diabetes and experimental models demonstrate loss from glomerular endothelial cells. Vascular endothelial growth factor (VEGF)A is upregulated in early diabetes, which is associated with albuminuria. Treatment with paracrine growth factors such as VEGFC, VEGF165b and angiopoietin-1 can modify VEGFA signalling, rescue albumin permeability and restore glomerular eGlx in models of diabetes. Manipulation of VEGF receptor 2 signalling, or a common eGlx biosynthesis pathway by these growth factors, may protect and restore the eGlx layer. This would help to direct future therapeutics in diabetic nephropathy.
Review overview
The focus of this review is to examine how components of the endothelial glycocalyx (eGlx) influence vascular permeability, particularly in the diabetic glomerulus, and the role they may have as a potential therapeutic target in diabetic nephropathy. First an overview of the eGlx will be given, followed by evidence demonstrating its importance in vascular permeability in health, diabetes and diabetic nephropathy. Finally, this review will present approaches that restore glomerular eGlx through common pathways, thereby highlighting therapeutic potential.
Endothelial glycocalyx (eGlx)
The eGlx is a complex carbohydrate meshwork, present on the luminal surface of blood vessels. It is primarily composed of glycoproteins (not a focus of this review) and proteoglycans, which have a protein core, embedded into the endothelial cell surface. Glycoprotein cores are attached to acidic oligosaccharides and terminal sialic acids, while proteoglycan cores are attached to multiple, long, unbranched glycosaminoglycan (GAG) chains [1]. The eGlx is often referred to as an extended endothelial surface layer (ESL) to incorporate the structural contribution of absorbed plasma proteins. It is a dynamic structure [2], with a high turnover rate, that is greatly influenced by surrounding factors.
Proteoglycan synthesis begins when ribosomes, bound to the endoplasmic reticulum (ER), synthesise a protein core backbone and feed it into the ER lumen. From here it is transported to the lumen of the Golgi apparatus where a serine residue of the core protein is attached to a tetrasaccharide via an O-glycosidic bond [3]. GAG chains are then extended from the non-reducing end by glycosyltransferase enzymes that alternately attach an amino and an acidic sugar [4–6]. The main core proteins found in the eGlx are transmembrane syndecans and membrane-bound glypicans [7], while the three main GAG are heparan sulphate (HS) chondroitin sulphate (CS) and hyaluronic acid (hyaluronan, HA). During polymerisation, GAG can be modified by sulfation (the addition of a sulfate group to an individual monosaccharide), which is what gives the eGlx an overall negative charge. GAG can also be modified by de-acetylation (removal of an acetyl group from a monosaccharide) and/or epimerisation (the alteration of a monosaccharide at one stereochemical position only e.g. in the transformation of glucuronic acid to iduronic acid) [8]. GAG distribution on core proteins is highly variable. For example, endothelial syndecans-1, -2 and -4 exhibit three GAG attachment sites, which predominantly bind HS. However, syndecan-1 has two additional sites specifically for chondroitin sulphate [9]. HA is a unique GAG as it is not sulphated, is unbranched, and is not linked to a core protein. It is the largest GAG with repeating disaccharide units reaching in excess of 106 Da [5]. These lengths are achievable as it is not constrained by the Golgi space during chain extension. It is synthesised at the inner surface of the plasma membrane by hyaluronic acid synthases (HAS1, HAS2 and HAS 3) [10].
Contribution of eGlx to microvascular permeability
Vascular permeability is the movement of water and molecules across the capillary wall and was originally modelled on Starling principles [11];
Vascular leakage and oedema are important concerns post-surgery with those experiencing perioperative ischemia (e.g. due to aortic clamping or cardiopulmonary by-pass). The importance of eGlx in vascular leakage in these patients was highlighted for the first time by Rehm et al. [19]. SDC1 and HS were measured in arterial blood of patients before, during and after vascular surgery. Both SDC1 and HS shedding were increased after different operative procedures in association with conditions of reperfusion (but not due to surgery itself) [19]. Using a complementary animal model of ischemia reperfusion, the authors confirmed that increased SDC1 and HS shedding were seen in association with reduced coronary microvascular eGlx depth, by electron microscopy, in guinea pig hearts.
Diabetes and eGlx
The eGlx has been shown to be altered in a number of pathological situations including ischemia-reperfusion injury and inflammation [20–22], hyperglycaemia [23], in chronic kidney disease [24], and during the progression of diabetes [25,26]. Diabetes is a major health concern worldwide. In the UK between 2015–2016, diabetes-related drugs made up to 10.6% of prescribed drugs used in primary care and 6.4% of individuals going to their GPs aged 17 yr and over were diagnosed with diabetes [27]. In general, diabetic patients suffer ill health, and there is strong correlation between diabetes and cardiovascular diseases [28,29]. More specifically, many of those with diabetes will suffer from complications associated with microvascular damage, which include retinopathy, neuropathy and nephropathy (collectively known as microangiopathy).
Diabetes and albuminuria
Diabetic nephropathy is the main cause of end stage renal failure in the western world [30]. It is considered a relatively late complication of diabetes, progressively occurring in susceptible patients 15–25 yr after the initial onset of diabetes [31,32]. Albuminuria is correlated with the progression of diabetic nephropathy [33] and can be categorised as normoalbuminuric (less than 30 mg/day), microalbuminuric (30–300 mg/day) or macroalbuminuria (300+ mg/day). It is also associated with endothelial dysfunction, decline in glomerular filtration rate (GFR) and increased risk of cardiovascular disease [34,35]. Of note, increased GFR also predisposes those with diabetes to the development of diabetic nephropathy [36]. Interestingly, even normoalbuminuria at the top end of the physiological range is an independent risk factor for cardiovascular disease in an otherwise health population [37–39].
Diabetes and eGlx dysfunction
Damage to the eGlx has been shown to occur in both Type-I and Type-II diabetes. Nieuwdorp et al. indirectly analysed the volume of the eGlx in patients with Type-I diabetes by imaging the erythrocyte-endothelium gap in sublingual microcirculation using side-stream darkfield imaging, before and after leukocyte passage [40]. Using this method, they demonstrated that; eGlx volume was reduced in Type-I diabetics; was exacerbated in those diabetics with microalbuminuria; and was correlated with increased blood plasma levels of HA and hyaluronidase (which cleaves HA). Indeed, in hyaluronidase knock out mice, the development of Type I diabetes-associated albuminuria was prevented [41]. EGlx volume has also been shown to be reduced in patients with Type-II diabetes in both the sublingual and retinal vascular beds. This was analysed using side-stream darkfield imaging and measuring the intravascular distribution of two fluorescent tracers of different sizes [26].
In addition to hyaluronidase, other enzymes have been implicated in GAG shedding in diabetes. For example, circulating matrix metalloproteinases (MMP) 2 and 9, known to cleave components of the eGlx [42], were significantly increased in Type I diabetic patients [43]. We suggest that TNF-𝛼, an inflammatory mediator in the diabetic milieu, induces MMP-9 activation, leading to HS cleavage from Sydecan-4 in glomerular endothelial cells [44]. Also, vascular endothelial growth factor (VEGF)A, which is upregulated in the diabetic milieu (discussed in more detail below), can induce glomerular MMP 9 expression [45]. Further to this, we have shown that, in an experimental model of albuminuria with glomerular eGlx dysfunction, MMP 2/9 inhibition restored glomerular eGlx and prevented albuminuria [46]. Another enzyme involved in GAG shedding in diabetes is heparanase (HPSE). Active HPSE degrades HS and is upregulated during diabetes in humans and in animal models [47–49]. Increased HPSE levels have also been demonstrated in the urine and plasma from Type II diabetic patients, and were found to positively correlate with increased glucose levels [48]. An increase in vascular permeability is also seen in mice that overexpress heparanase [50] and this enzyme has been further linked to the pathophysiology of childhood steroid-sensitive nephrotic syndrome [51].
Hyperglycaemia, an important mediator of the diabetic milieu in Type I and II diabetes, is thought to impact eGlx health. Nieuwdorp et al. showed that acute induction of hyperglycaemia in healthy individuals reduced eGlx volume within 6 hours [52]. In culture, hyperglycaemia inhibits synthesis of sulphated and non-sulphated GAG, with no demonstrable change in proteoglycan expression [53]. Together, these studies demonstrate that eGlx damage is strongly associated with the development of albuminuria in diabetes, indeed, the eGlx has been proposed as the site of initial damage leading to microalbuminuria [54]. Therefore, the eGlx may be a promising target to reduce albuminuria and protect against other microvascular complications.
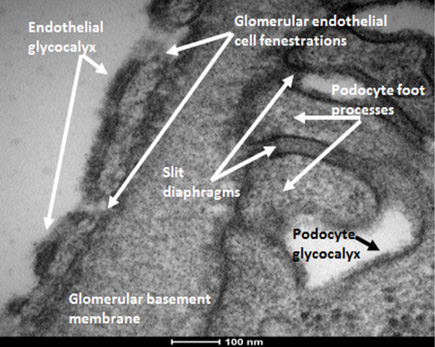
Transmission electron micrograph of the glomerular filtration barrier showing endothelial glycocalyx. Representative image from an FVB strain mouse, anaesthetised and perfusion fixed with Glutaraldehyde containing the cation, Alcian blue. This enabled visualisation of the electron dense, negatively charged glycocalyx on GEnCs. Of note, podocytes are also coated in glycocalyx.
Understanding how the glomerulus regulates filtration and maintains selective (restricted) protein passage is key to understanding the development and progression of renal pathologies in which this restriction is lost, such as in diabetic nephropathy. Blood is filtered across the glomerular capillary through a highly specialised filtration barrier. This multi-layered structure prevents large proteins, such as albumin, from leaving the blood whilst allowing minimally restricted movement of water and small solutes. Traditionally the glomerular filtration barrier (GFB) is presented as a three-layer structure consisting of podocytes (epithelial cells), the glomerular basement membrane (GBM) and fenestrated glomerular endothelial cells (GEnC) [55]. However, the glomerular eGlx, highlighted in Fig. 1, has also been shown to significantly contribute towards the GFB [56]. Tissue culture studies show that GEnC synthesise and express HS [57], CS and HA [58]. CS and HA can be seen within human glomeruli in Fig. 2. In the glomerulus, HS has been identified not only in the eGlx but also in the GBM where it is one of the 4 main components along with laminin, collagen IV and nidogen. Here it is primarily found as part of the proteoglycan, agrin [59,60]. Whether GBM HS plays a role in protein restriction is under debate [59,61–63], although a general note for these and all Glx studies is that there is a need to discriminate between GAG of eGlx and elsewhere (i.e GBM, podocytes and mesangial cells).
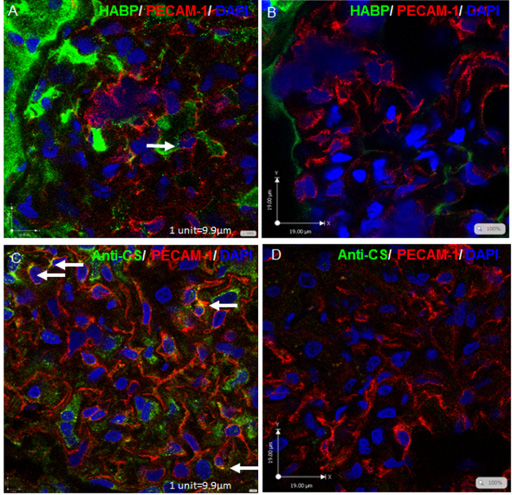
Hyaluronic acid and chondriotin sulphate expression in human glomeruli. Fresh frozen sectioned human glomeruli were treated with 250 mg/ml HYAL (B), 100 mU/ml chondroitinase (D), to show specificity, or left untreated (A and C). They were then fixed and immunostained with biotinylated hyaluronic acid binding protein (HABP) in conjunction with strepavidin-AF488(green) and anti-PECAM-1 (red) (A and B), or anti-CS in conjunction with anti-mouse IgM AF488 (green) anti-PECAM-1(red) (C and D), and counterstained with DAPI (blue). Confocal Z stacks are shown. Architecture is not optimal in fresh frozen tissue and capillary loops are not apparent. Arrows indicate co-localisation of GAG with GEnCs, suggesting eGlx.
The glomerular eGlx has been suggested to restrict both solutes and fluid (not just large, charged proteins) according to a mathematical simulation of the GFB [64]. Its contribution to water permeability was confirmed experimentally when a bolus of hyaluronidase or heparinase, given in vivo for up to an hour, led to an increase in GFR [65]. EGlx damage in experimental models of diabetes was associated with increased glomerular hydraulic conductivity (a measure of single nephron GFR). Further, treatment that restored eGlx depth also rescued hydraulic conductivity [66]. In cultured human GEnC, targeted eGlx removal led to an increase in trans-endothelial electrical resistance, indicating increased water and solute flux [57,58].
Various studies have highlighted the importance of eGlx in glomerular large solute (i.e. albumin) permeability. For example, glomerular eGlx removal, by hypertonic saline in rats, caused an increase in fractional albumin clearance by 12-fold [67]. Additionally, both hyaluronidase and chondroitinase individually increased the fractional clearance of albumin in isolated, perfused, mouse kidneys ex vivo (although not in vivo) [65,68]. This was associated with reduced glomerular eGlx depth, although measurements were indirect. However, it is worth noting that chondroitinase and hyaluronidase can digest HA and CS respectively, albeit at a slower rate [69,70]. Hyaluronidase given over 4 wk in mice reduced glomerular eGlx coverage and caused an increase in albumin passage across the GFB (but not urinary albumin creatinine ratio (uACR)) [71]. This was determined by anti-albumin immunogold staining in the GFB. We have recently shown that a low dose combination of hyaluronidase and chondroitinase, given acutely (30 min) or chronically (over 2 wk), reduced eGlx coverage without off-target effects [72,73]. Under the same conditions we directly measured changes in glomerular albumin permeability, which was significantly increased following enzyme treatment. By measuring glomerular albumin permeability directly, we could bypass tubular reabsorption of albumin which impacts on the sensitivity of uACR measurements [72]. In vitro, low dose, targeted enzymatic removal of CS, HS, and HA on cultured GEnC (quantified by immunofluorescence) demonstrated that removal of CS and HS, but not HA, increased albumin passage across GEnC monolayers [57,58]. These treatments demonstrate that specific removal of eGlx leads directly to a rapid and sustained increase in glomerular albumin permeability.
Other non-enzyme-based studies have also demonstrated how the eGlx contributes towards GFB function. Aged Munich-Wistar Frömter (MWF) rats with spontaneous proteinuric kidney disease have decreased eGlx and increased glomerular albumin permeability [74]. Additionally, intravenous administration and subsequent absorption of wheat germ agglutinin (WGA) lectin decreased glomerular albumin permeability in aged rats, indicating that modifications to the eGlx can improve glomerular function [74]. Genetic deletion of the linker between HS chains and perlecan (a core protein) caused mice to become proteinuric [75]. We have recently shown that aldosterone, in combination with high salt, damages glomerular eGlx in vivo, leading to an increased glomerular sieving coefficient of albumin [46]. This follows from interesting work by Oberleithner et al. on salt overload and eGlx stiffness. Atomic force microscopy studies demonstrated that salt overload increased eGlx stiffness and reduced eGlx depth in cultured endothelial cells in an aldosterone dependent manner [76], a potential mechanism for increased sodium accumulation in the tissues. These experiments highlight the importance of the eGlx in preventing excess protein passage.
EGlx restoration and diabetic nephropathy
The key question is; how can we restore eGlx? Significant progress has been made in directly targeting the eGlx in diabetic patients. Sulodexide, a mixture of low-molecular weight GAG (heparan (80%) and dermatan sulfate (20%)), has been used to treat microvascular complications in diabetic patients [77]. Initially, several small scale clinical studies demonstrated successful, therapeutic use of Sulodexide, showing an increase in sublingual eGlx depth (using side-stream darkfield imaging), a decrease in albuminuria (most effective in those with microalbuminuria), and slowed progression of diabetic nephropathy [26,78]. Unfortunately, two larger randomized double-blinded placebo control studies later demonstrated that treatment with Sulodexide did not reduce albuminuria, therefore the need for a new and effective treatment remains [77,79]. However, it should be noted that many consider the effects of Sulodexide to have been undervalued in these later studies [80–83]. Overall, these data suggest that the eGlx may be amenable to therapeutic intervention in diabetes and that targeting this structure could prove the key for treating this disease. Experimentally, restoration of the eGlx has been demonstrated to be achievable and effective in blood vessels outside of the kidney in the coronary [84], pulmonary [85] and mesenteric microcirculation [16]. We have also demonstrated that the eGlx can be restored in glomeruli [46,66,86] which will be key in targeting albuminuria.
Glomerular vascular endothelial growth factors (VEGF)
Podocyte-GEnC crosstalk is critical to maintain normal glomerular function. Growth factors form a major part of this communication, but during disease states, these can become imbalanced. One example of these is the VEGF family of proteins. In humans there are five VEGF variants, VEGF-A, -B, -C, -D and placenta growth factor [87]. These proteins each have distinct biological roles and are important in blood and lymph vessel formation and vascular homeostasis. Alternative splicing of the 8 exon, 7 intron VEGF-A gene gives rise to a number of mature isoforms with different biological activities. Of these, VEGF165 is the most commonly found variant throughout the body [88,89]. VEGF165 is important for GEnC fenestration formation and health via its main signalling receptor, VEGF receptor 2 (VEGFR2) [90–92]. However, transgenic overexpression of VEGF165 can cause increased GFR in the short term, followed by loss of GFR in the long term [93]. In addition, podocyte-specific overexpression of VEGF165 was shown to induce albuminuria, increase glomerular VEGFR2 phosphorylation and increase glomerular MMP 9 expression [45]. Additionally, VEGF165 has also been shown to be upregulated in early diabetic nephropathy in human disease [94,95] and in experimental models [96–99]. VEGFR2 was also shown to be upregulated in early experimental diabetic nephropathy and was associated with increased GEnC VEGF165-VEGFR2 signalling [96]. This has been shown to cause loss of permselectivity and albuminuria [100]. Of note, VEGF165 may also induce eGlx loss. As well as promoting glomerular MMP 9 expression [45], it also promoted ADAM17 (another member of the MMP family of proteins) activation in cultured human GEnC [58]. Furthermore, VEGF165 has been shown to increase shedding of sulphated GAG in cultured human GEnC [58]. In the following section, paracrine growth factors will be described that may counterbalance the detrimental effects of glomerular VEGF165 to protect from albuminuria in diabetes. We suggest they may act by antagonising VEGF165/VEGFR2 signalling to restore the glomerular eGlx. Of course, VEGF165 signalling is just one mediator of diabetic nephropathy. As well as counterbalancing the effects of the VEGF axis, we believe the end point of these paracrine growth factors, restoring eGlx, would go some way to counteract the generic effects of the diabetic milieu.
Podocyte paracrine growth factors
i. VEGF165b: In 2002, Bates and colleagues described a novel family of VEGFA isoforms, where the last 6 amino acids differ due to alternative splicing of exon 8, such that the terminal end of exon 8 is transcribed instead of the proximal end. These are depicted as VEGFxxxb. For example, VEGF165b has been shown to bind VEGFR2 with the same affinity as VEGF165 [101]. Podocyte-specific VEGF165b expression in mice has been shown to block the increase in glomerular hydraulic conductivity induced by VEGF165 [102]. Reduced glomerular hydraulic conductivity has also been demonstrated in glomeruli isolated from diabetic mice, rats and humans treated ex vivo with VEGF165b [66]. Further, in a mouse model of Type I diabetes, podocyte-specific expression of VEGF165b significantly reduced albuminuria [66]. This was also achieved when a dual insult of VEGF165 overexpression was induced alongside the induction of diabetes, supporting the hypothesis that VEGF165b reduces albuminuria through antagonising VEGF165. Recombinant human VEGF165b protected against albuminuria in a Type II model of diabetes, but only when administered before the development of albuminuria. Thus, VEGF165b appears to have an impact on water/small solute and large solute permeability in the GFB. The protective effects of VEGF165b on glomerular hydraulic conductivity in diabetic rats was shown to be VEGFR2 dependent [66]. The activation of VEGFR2 by VEGF165b appears to be endothelial cell-type dependent. In human umbilical vein (large vessel) endothelial cells, VEGF165b could bind, but not phosphorylate, VEGFR2 yet in human microvascular endothelial cells it resulted in the phosphorylation of Akt [101]. Of note, overexpression of VEGF165b in glomeruli caused increased VEGFR2 expression and phosphorylation in both healthy and diabetic mouse glomeruli [66]. This suggests that VEGF165b could have differential VEGFR2 signalling effects in different vascular beds. Critically, VEGF165b has also been shown to modify the eGlx. This was demonstrated in an early model of Type I diabetes in mice where human recombinant VEGF165b administration restored glomerular eGlx depth. This may be via delayed downstream signalling by VEGF165b-induced activation of VEGFR2, potentially involving a role for VEGFR2/VEGFR1 heterodimer formation [103].
ii. Ang1: Angiopoietins are a family of endothelial cell growth factors, with essential roles in regulating vascular growth, development and maturation. The complex interaction between the angiopoietins and vascular endothelial growth factors determine endothelial cell behaviour, therefore they are key players in microvascular permeability, both in health and diabetic nephropathy [104–107]. The two major members of the angiopoietin family are angiopoietin-1 (Ang1) and angiopoietin-2 (Ang2) [108]. Tie2 is the main signalling receptor and binds both Ang1 and Ang2, although in most circumstances only Ang1 induces receptor phosphorylation [109–111]. Ang2 predominantly antagonises Ang1-induced phosphorylation of Tie2 [110]. Tie2 together with VEGFR2 activation is required for endothelial cell survival and vessel stabilization [92,112,113]. In healthy mice, Ang1 reduced the vascular leakage of 70 kDa macromolecules from newly formed mouse blood vessels [114] and from mature vessels (mouse and human) following treatment with permeabilising reagents [115,116]. Moreover, Ang1 reduced hydraulic conductivity in rat glomeruli and in continuous frog mesenteric vessels [16]. The reflection coefficient (a measure of macromolecular restriction) in continuous vessels was also increased. Critically, Ang1 increased eGlx depth in continuous vessels. All these data indicate that Ang1 decreases vascular water and solute permeability, potentially via eGlx. Therefore, we hypothesised that Ang1 could modify glomerular eGlx to restrict albuminuria.
Of note, Ang-2 reduced eGlx depth in human umbilical vein endothelial cells in a heparanase dependent manner, which was blocked by Ang-1 [117]. Further, in an in vivo model, Ang-2 was shown to increase vascular leak which was blocked by heparanase inhibition [117]. In diabetes the balance between Ang1 and Ang-2 is lost, which also impacts on VEGF165. Ang2 mRNA was upregulated in human diabetic glomeruli and Ang1 was significantly decreased in a Type 1 diabetic mouse model [104]. In these mice, when Ang1 was overexpressed by podocytes, albuminuria was reduced and Tie2 phosphorylation was increased, highlighting the protective effects of Ang1 in diabetes. Of note, in the diabetic mice, glomerular VEGFA expression was increased and glomerular VEGFR2 expression was decreased, but VEGFR2 phosphorylation was increased. Notably, podocyte-specific overexpression of Ang1 significantly reduced the increased VEGFR2 phosphorylation [104]. Also, kidney cortex soluble VEGFR1 expression was increased, which can sequester VEGFA. Therefore, the effects of Ang1 may be two-pronged; Tie2 activation and VEGF165 signalling suppression.
Our group investigated if the protective capabilities of Ang1 in diabetes may be mediated by the eGlx. First, we confirmed that Ang1 (given ex vivo) significantly reduced albumin permeability in glomeruli isolated from Type 1 diabetic rats [72]. We then demonstrated that recombinant Ang1, given i.v. 30min before sacrifice, significantly restored glomerular eGlx in diabetic animals, shown by quantitative electron microscopy. This suggests that Ang1 effects glomerular function through glomerular eGlx [72].
iii. VEGFC: VEGFC is most commonly associated with developmental lymphangiogenesis, where it is required for normal development of the endothelial lymphatic system, signalling through VEGFR3 [118]. It also signals through VEGFR3 to maintain the differentiated lymphatic endothelium in adults [119]. Like VEGFA, VEGFC can signal to vascular endothelial cells, increasing migration and proliferation by stimulating VEGFR2 [120]. Binding to VEGFR2 by VEGFC occurs at the same binding site as VEGF165, as indicated by the ability of each growth factor to displace the other [120]. In the glomerulus, VEGFC is expressed by podocytes and acts on GEnC [121]. Unusually, we could not demonstrate phosphorylation of VEGFR3 by VEGFC in cultured human GEnCs, but could show phosphorylation of VEGFR2, albeit over a longer time course than VEGFA (30 min v.s. 2 min) [121]. We examined phosphorylation of VEGFR2 tyrosine residues by VEGFC, however we did not find significant phosphorylation at Tyr 951, 1175 or 1214; sites involved in VEGF165 activation and downstream signalling (Fig. 3). This suggests that VEGFR2 phosphorylation is delayed and perhaps total phosphorylation is reduced in GEnC by VEGFC. In these cells VEGFC reduced macromolecular protein passage [121]. We suggest that this is due to the synthesis of unsulfated and sulphated GAG by VEGFC (discussed in more detail below) [122]. Using our glomerular albumin permeability assay, we demonstrated that ex vivo treatment of isolated mouse glomeruli with VEGFC blocked the increased permeability induced by VEGF165. In addition, VEGFC could rescue raised glomerular albumin permeability in glomeruli isolated from Type II diabetic (and albuminuric) mice [73]. We have also demonstrated that the effects induced by VEGFC, like the other protective growth factors discussed, may be mediated via the eGlx. Glomerular albumin permeability was increased in mice administered GAG shedding enzymes, both acutely (30 min) and chronically (2 wk), but VEGFC blocked this effect whilst restoring eGlx depth and/or coverage [73].
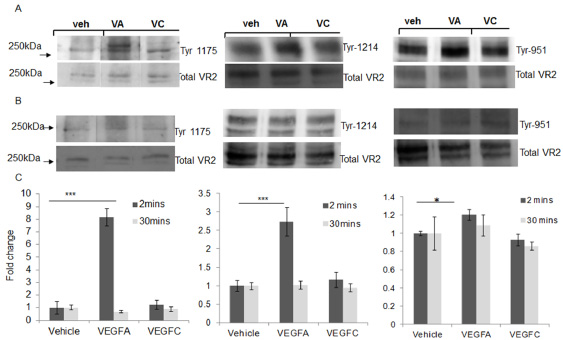
VEGFC does not induce VEGFR2 phosphorlyation at tyrosine (ty) sites 1175, 1214 or 951 in cultured glomerular endothelial cells (GEnC). GEnC were stimulated with 1 nM VEGFA (VA), 10nM VEGFC (VC) or vehicle (Veh) for 2 min (A) or 30 min (B). Cells were lysed and samples subjected to Western blotting for specific VEGFR2 tyrosine residues ty1175, ty1214 or ty951 and total VEGFR2. Representative blots are shown. Densitometry summary data is shown in C. One-way ANOVA, Bonferroni post hoc tests indicated. ∗ = p < 0.05, ∗∗∗ = p < 0.001, n = 3–6.
We went on to investigate the effect of VEGFC on albuminuria and eGlx in vivo using podocyte-specific overexpression of VEGFC. Of note, VEGFC reduced glomerular VEGFA mRNA expression and had no long term detrimental effects on glomerular function [73], in contrast to that described for podocyte-specific overexpression of VEGFA [45]. Glomerular VEGFR2/VEGFR3 heterodimer formation was significantly increased in podocyte-specific VEGFC overexpressing mice [73]. This suggests that VEGFC may antagonise VEGF165 signalling by competitively binding VEGFR2, but may induce differential signalling through modification by VEGFR3. We confirmed that podocyte-specific overexpression of VEGFC could rescue albuminuria in a Type I diabetic mouse model, if it was expressed before albuminuria developed [73]. Increased glomerular VEGFR2 expression in diabetic mice was significantly reduced by VEGFC, supporting our hypothesis that VEGFC antagonises VEGF165/VEGFR2 signalling. Together, this data suggests that VEGFC protects from albuminuria through modification of VEGFR2 signalling and eGlx restoration.
VEGF165b, Ang1 and VEGFC all protect from increased glomerular VEGF165 signalling and from albuminuria in diabetes, preferentially when administered during early development. Critically, these growth factors counterbalance VEGFA signalling, either through expression/sequestering of VEGF165 and/or potential modification of VEGFR2 signalling. Blockade of the glomerular VEGF165 system in disease needs to be moderate to avoid additional glomerular dysfunction [92,123–125]. Perhaps counterbalancing by these important endogenous mediators provides the key. In addition, restoration of the glomerular eGlx by these growth factors was associated with reduced glomerular small solute (VEGF165b and Ang1) and albumin permeability (VEGF165b, Ang1, VEGFC). We know that VEGF165b restores glomerular eGlx and that this leads to increased WGA lectin binding [66]. WGA binds N-acetyl glucosamine, a component of HS and HA, and N-acetyl neuraminic acid, which is a major sialic acid. This gives some clues as to the components of the VEGF165b-restored eGlx. More extensive investigations have been carried out for Ang1 and VEGFC. Ang1 can restore/enhance eGlx in frog mesentery vessels and glomerular capillaries in vivo within 30 min. Whether this is due to de novo synthesis or not has yet to be proven, but inhibition of vesicle translocation by Brefeldin A blocked the effect, indicating that restoration may come from pre-existing pools of GAG [16]. In human microvascular endothelial cells in vitro, Ang1 increased sulphated GAG content of cell supernatant [16]. GAG were also shown to be effected by Ang-1 in cultured human GEnC, where treatment induced de novo synthesis of both sulphated and un-sulphated GAG, as determined by 3H glucosamine and liquid chromatography, within 1 hr (Desideri et al., unpublished). These experiments suggest that rapid de novo synthesis in vivo is a plausible explanation. Using the same technique, de novo GAG synthesis was also seen in GEnC in response to VEGFC after 24 hr [58]. 76% of sulphated GAG synthesised by VEGFC were highly sulphated, compared to only 32% under control conditions. Additionally, using immunofluorescence, VEGFC was shown to induce HA and CS synthesis. Furthermore, VEGFC significantly increased NDST2 mRNA, a sulfotransferase, which supports observations of increased eGlx charge. As mentioned previously, these effects by VEGFC are in contrast to VEGF165, which may promote GAG shedding [45,58]. This leads to a complex interaction between these families of growth factors, VEGF165/VEGFR2 signalling and eGlx modifications as summarised in Fig. 4.
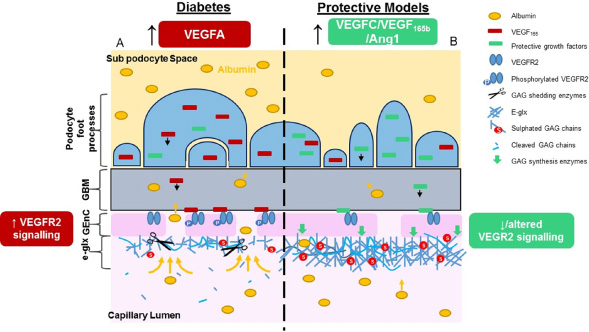
The effect of protective paracrine growth factor signalling on restoration of eGlx in the glomerular filtration barrier. VEGF165 podocyte overexpression in early diabetic nephropathy leads to enhanced glomerular endothelial VEGFR2 activation, a damaged eGlx and increased albuminuria (A). The overexpression of paracrine growth factors VEGF 165b, Ang1 and VEGFC can impact the detrimental signalling of VEGF165 in a number of ways; podocyte VEGF165 expression or availability may be reduced; VEGFR2 signalling may be reduced/altered; enzymes associated with the synthesis, modification or shedding of GAG may be rebalanced such that the eGlx layer is restored and albuminuria is reduced.
In conclusion, there are similarities arsing in the mechanism of protection of these paracrine growth factors. The next step is to identify the common pathways influenced by these growth factors that promote restoration of glomerular eGlx, and those that protect from degradation. This will prove to be a powerful, multi-targeted approach to albuminuric diabetic nephropathy. In addition, eGlx restoration by the same means has the potential to treat other microvascular disorders where capillary leak to small solutes and large proteins is a key step in the progression of the disease.
List of abbreviations
Ang-1
Angiopoietin-1
A disintegrin and metalloproteinase domain 17
ADAM17
Chondroitin sulphate
CS
eGLX
Endothelial glycocalyx
Endoplasmic reticulum
ER
Endothelial surface layer
ESL
GAG
Glycosaminoglycans
Glomerular endothelial cells
(GEnC)
Glomerular basement membrane
GBM
Glomerular filtration barrier
GFB
Glomerular filtration rate
GFR
Hyaluronidase
HYAL
Hyaluronic acid
HA
Hyaluronic acid synthase
HAS
Heparanase
HPSE
Heparan sulphate
HS
Hydroxyethylstarch
HES
Matrix metalloproteinase
MMP
Munich Wistar Frompter
MWF
Urine albumin creatinine ratios
uACR
Vascular endothelial growth factor
VEGF
Vascular endothelial growth factor receptor
VEGFR
Wheat germ agglutinin
WGA