
Abstract
BACKGROUND:
Non-small lung cancer ranks first in the cancer-related death of all malignant tumors. Exploring novel biological targets is of great significance for diagnosis and therapy of NSCLC.
OBJECTIVE:
In this study, we aimed to explore the effect of LINC00668 on the biological functions of NSCLC cells and the underlying mechanism.
METHODS:
RT-qPCR assays and western blot assays were utilized to estimate the relative gene expression at mRNA and protein levels, respectively. CCK8, colony formation, wound healing, transwell, and cell apoptosis assays were employed to assess cell function. IHC and FISH assays were used to determine the gene expression in NSCLC tissues. RIP and dual-luciferase assays were conducted to validate the combination between LINC00668 and miR-518c-3p. The correlation of expression between miR-518c-3p and LINC00668 or TRIP4 was determined by Pearson correlation analysis.
RESULTS:
LINC00668 was aberrantly upregulated in NSCLC tumor tissues and cell lines. Inhibition of LINC00668 significantly suppressed tumor proliferation, migration, invasion and promoted cell apoptosis. Mechanistically, LINC00668 could bind to miR-518c-3p, thus targeting the 3’UTR of TRIP4. TRIP4 overexpression rescued the weakened cell function mediated by LINC00668 silencing.
CONCLUSIONS:
LINC00668 acted as an oncogene in NSCLC progression through miR-518c-3p/TRIP4 axis. Our study disclosed a new mechanism of LINC00668 functioned in NSCLC and may give a deeper insight of the targeted therapy of NSCLC in the future.
Introduction
Lung cancer is one of the major death-caused malignancies that threaten the global health. According to the 2020 global cancer statistics, there were approximately 2.2 million new cases of lung cancer, ranking second, and 1.8 million deaths, ranking first [1, 2]. Non-small cell lung carcinoma (NSCLC) accounted for the majority of all histological subtypes of lung cancer [3, 4]. Regrettably, the five-year overall survival rate for NSCLC was less than 25% for all stages combined, underscoring the poor prognosis for affected individuals [5, 6]. Thus, identifying novel molecular targets for clinical diagnosis and treatment, and improving NSCLC patient prognosis were of paramount significance.
Long non-coding RNAs (lncRNAs) comprised a group of RNAs that exceed 200 nucleotides in length and lacked the capacity for protein coding or could only encode some small peptides [7, 8]. LncRNAs were categorized based on their relative location to protein-coding genes, including long intergenic non-coding RNAs (lincRNAs), natural antisense transcripts (NATs), bidirectional lncRNAs, overlapping transcripts, and sense intronic lncRNAs[9]. They participated in tumorigenesis and malignant tumor progression through various mechanisms, including chromatin modification, miRNA sponging, endogenous siRNA formation, and promoter activation [10, 11]. Recent evidence has confirmed that lncRNAs play crucial roles in the epigenetic modification of NSCLC. For example, LINC00673 sponged miR-150-5p to regulate ZEB1 expression, a key regulator of epithelial mesenchymal transition [12]. LY6K-AS promoted mitotic progression by regulating H3 lysine 4 trimethylation (H3K4me3) transcription at the promoter of kinetochore members [13]. MELTF-AS1 promoted NSCLC tumorigenesis by binding to YBX1 and modulating the activation of ANXA8 [14].
LINC00668, an oncogene in the development of numerous malignant tumors, has been identified to exhibit high expression levels and poor prognosis. For instance, LINC00668 was upregulated in breast cancer and promoted cell invasion and metastasis by interacting with SND1 [15]. LINC00668 sponged miR-118-5p and regulated USP47 expression to promote tumorigenesis and progression in colorectal cancer [16]. LINC00668 facilitated cell metastasis by binding to HuR-dependent upregulation of PKN2 in gastric cancer [17]. Nevertheless, the functional roles and underlying mechanisms of LINC00668 in NSCLC have not been thoroughly investigated. In this study, we validated the high expression of LINC00668 in NSCLC cell lines and performed loss-of-function assays, which indicated that LINC00668 promoted NSCLC progression. Through dual-luciferase report assays and RNA immunoprecipitation assays, we observed that LINC00668 sponged miR-518c-3p to competitively regulate the downstream gene TRIP4. Our findings suggested that the LINC00668/miR-518c-3p/TRIP4 axis played a pivotal role in the oncogenic progression of NSCLC. LINC00668 was potential to become a diagnostic and therapeutic biomarker in the future.
Materials and methods
Cell culture and transfection
The human lung epithelial cell line Beas-2B (CL-0496) and NSCLC cell lines H1703 (CL-0390), SK-MES-1 (CL-0213), H520 (CL-0402) were procured from Procell in Wuhan, China. A549 (SCSP-503), H1299 (SCSP-589), and H1975 (SCSP-597) were purchased from the cell bank of Chinese Science Academy in Shanghai, China. SPCA1 (CL0296) was obtained from Huifeng in Hunan, China. A549, H1299, and SK-MES-1 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Rockville, USA) supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin (Gibco, Rockville, USA), while the Roswell Park Memorial Institute (RPMI-1640) medium (Gibco, Rockville, Maryland, USA) with the same components was used to culture SPCA1, H1975, H1703, H520, and Beas-2B. All cell lines were confirmed to be free of mycoplasma infection using MycoBlue Mycoplasma Detector (Vazyme, Nanjing, China), and cultured in a controlled environment of 37
RNA extraction and quantitative real-time PCR (qRT-PCR)
The total RNA was extracted using RNAiso Plus (Invitrogen, Carlsbad, CA, USA) in a sequential manner involving trichloromethane, isopropanol, and ethanol. Subsequently, cDNA was reversely transcribed from the extracted RNA using the PrimeScript
Fluorescence in situ hybridization assays (FISH)
Briefly, the fresh normal and cancerous lung tissues were processed according to the instructions of the in-situ hybridization kit (Beyotime, R0306S) after fixation, gradient dehydration, embedding, and sectioning. The final sample was imaged under inverted fluorescence microscope (Nikon, Ts-2R) after acetylation, prehybridization, probe denaturation, hybridization, and nuclear staining.
Cell proliferation assays
The cell proliferation was estimated by cell counting kit-8 assays and colony forming assays. Specifically, 5
Cell apoptosis assay
After 48 hours of transfection, the supernatant containing the suspended cells was collected first, followed by the addition of the digested adherent cells to the corresponding flow cytometry tube. Apoptosis of the cells was assessed using the Annexin V-FITC Apoptosis Detection Kit (eBioscience, San Diego, USA), according to the manufacturer’s instructions. In brief, each sample was treated with 5
Cell migration and invasion assays
The wound-healing assays were carried out to assess cell migration. In brief, cells were seeded into 6-well plates and incubated until they fully covered the well surface. Then, a 200
Western blot assay
To extract proteins, RIPA lysis buffer (Beyotime, Shanghai, China) was used, and protein degradation during lysis was inhibited using protease inhibitor cocktail (Millipore Sigma, Darmstadt, Germany). The protein concentration was determined using the BCA Protein Assay kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Proteins were separated by 4%–20% SurePAGE (GeneSript, Nanjing, Jiangsu, China) under constant voltage electrophoresis, and the protein gel was transferred to a PVDF membrane (Millipore, Massachusetts, USA). After the transfer, the membrane was blocked with 5% bovine serum albumin (BSA) in TBST for one hour. Subsequently, the membrane was incubated with GAPDH antibody (1:5000) and TRIP4 antibody (1:1000) overnight. These antibodies were purchased from Zen-bio, Suzhou, China. Following primary antibody incubation, HRP Goat Anti-Rabbit IgG (Beyotime, Shanghai, China) at a dilution ratio of 1:5000 was used for secondary antibody incubation. The band was then developed using Tanon ECL (Shanghai, China) and detected under Tannon 5200 (Shanghai, China) using gray scale analysis by ImageJ Software.
Dual-luciferase report assay
The pGL6-miR-linc00668-wt, pGL6-miR-linc00668-mut, pGL6-miR-TRIP4-wt, and pGL6-miR-TRIP4-mut plasmids were synthesized by General biosystems, North Carolina, USA. The pRL-TK plasmid was utilized as a renilla luciferase reporter. Co-transfection of miR-518c-3p NC (or mimic) and the wild-type plasmid (or mutant type) was performed using Lipofectamine 3000 (Invitrogen, Tokyo, Japan). Luciferase assays were conducted using the Dual-Luciferase
RNA Immunoprecipitation (RIP) assays
RIP assays were performed using the EZ-Magna RIP kit (Millipore, MA, USA) following the manufacturer’s instructions. The IgG antibody (Abcam, Cambridge, UK) and AGO2 antibody (Proteintech, Rosemount, USA) were combined with Magnet Protein A/G beads (Invitrogen, Carlsbad, USA), and incubated with cell lysates of H1299 and A549. Following this, the protein was digested by Protein K at 55
Immunohistochemistry (IHC) staining
The fresh tissues were fixed in 4% paraformaldehyde for over 24 h and then dehydrated using graded ethanol. Following, the tissue soaked in paraffin was embedded in an embedding machine and cut into sections at 4
Statistical analysis
The data between different experimental groups were tested for normality, and the statistical differences were observed using the unpaired student
Results
LINC00668 was upregulated in NSCLC and LINC00668 knockdown suppressed biological behaviors of NSCLC cells
LINC00668 was aberrantly upregulated in NSCLC and LINC00668 knockdown suppressed cell proliferation, migration, invasion, and facilitated cell apoptosis in NSCLC cell lines. A. LINC00668 was upregulated in NSCLC cell lines (A549, SPCA1, H1975, H1703, SK-MES-1, and H520) compared with Beas-2B; B. LINC00668 was upregulated in LUAD and LUSC tissues in the GEPIA database; C. LINC00668 expression was upregulated in NSCLC tissues compared to normal tissues using FISH assays; D. LINC00668 knockdown impaired the cell viability of A549 and H1299 cells through CCK8 assays; E. Suppression of LINC00668 decreased the cell colonies; F. LINC00668 inhibition accelerated cell apoptosis in NSCLC cells; G–H. LINC00668 silencing inhibited cell invasion (G) and migration (H) of A549 and H1299 cells using transwell assays and wound healing assays respectively. Data were analyzed by unpaired student’s 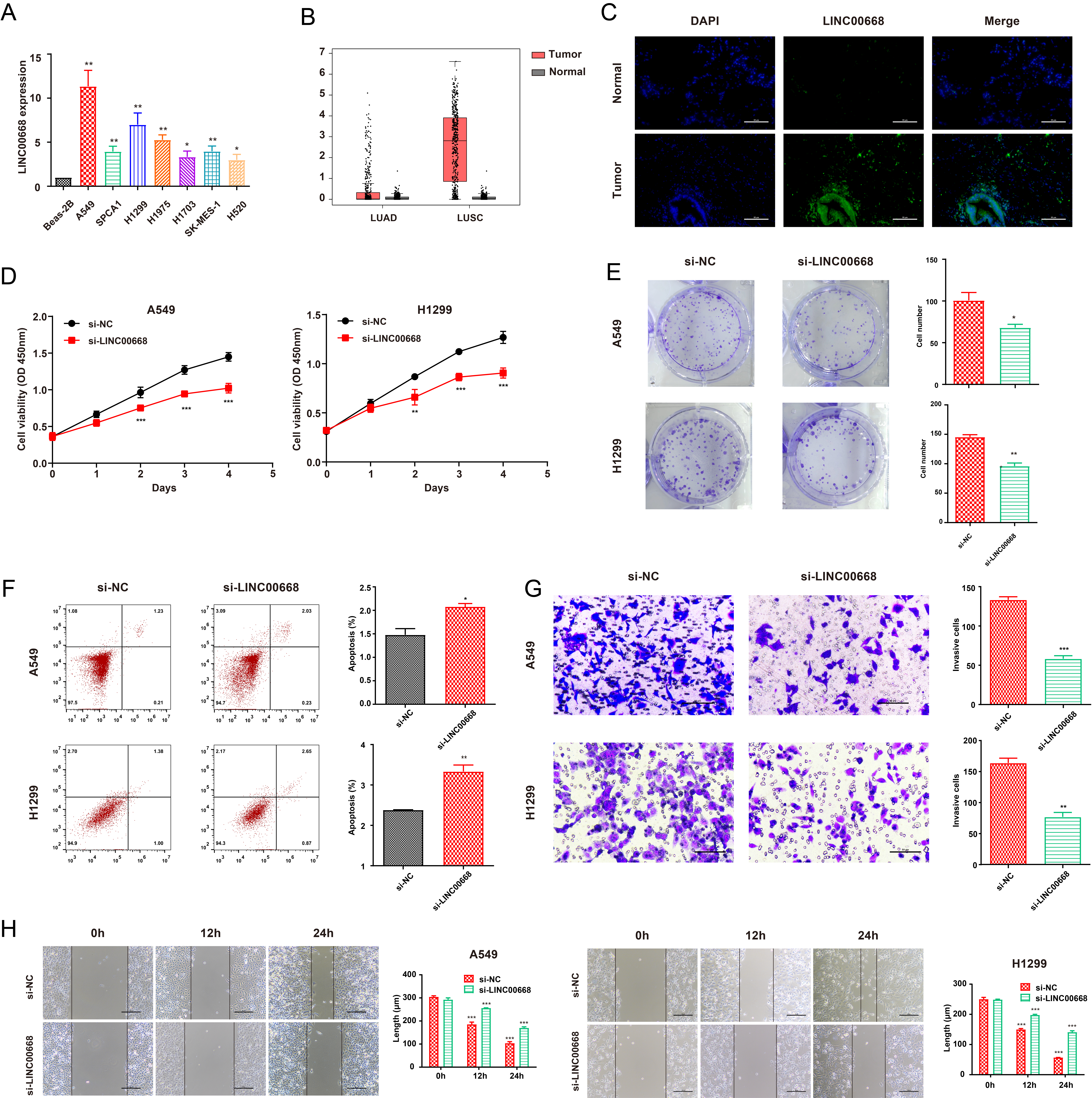
We first detected the differential expression level of LINC00668 in NSCLC cell lines compared with the normal. The qRT-PCR assays were performed, and the results demonstrated a significant upregulation of LINC00668 expression in A549, SPCA1, H1299, H1975, H1703, SK-MES-1, and H520 compared to normal pulmonary epithelial cells Beas-2B (Fig. 1A). A549 and H1299 showed the highest expression of LINC00668 and were selected for subsequent experiments. LINC00668 was upregulated in lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) compared to normal tissues according to the GEPIA database (Fig. 1B). Through FISH assays, we found there were positive signals after hybridization in tumor lung tissue in contrast to normal lung tissue (Fig. 1C).
In order to examine the biological function of LINC00668, a specific small interfering RNA (siRNA) was designed to silence LINC00668 expression. Subsequently, knockdown of LINC00668 led to a decrease in cell viability and colony formation in NSCLC, as shown in Fig. 1D and E. Moreover, cell apoptosis assays showed an increase in the rate of apoptosis in NSCLC cell lines following LINC00668 silencing, as depicted in Fig. 1F. Additionally, transwell assays suggested that LINC00668 knockdown restrained the invasion capability of A549 and H1299 cells (Fig. 1G). Furthermore, wound healing assays indicated that the downregulation of LINC00668 inhibited the migration of NSCLC cells (Fig. 1H). Overall, these findings suggested that LINC00668 silencing suppressed cell proliferation, invasion, and migration while accelerating cell apoptosis in NSCLC.
LINC00668 sponged miR-518c-3p and knockdown of LINC00668 upregulated miR-518c-3p expression in NSCLC. A. miR-518c-3p had a binding locus for LINC00668 based on Starbase; B. miR-518c-3p was downregulated in 7 NSCLC cell lines (A549, SPCA1, H1299, H1975, H1703, SK-MES-1, and H520) compared with Beas-2B; C. Dual-Luciferase assays detected the luciferase activities after being co-transfected with miRNA (NC or miR-518c-3p) and luciferase reporter vectors (WT or MUT); D. miR-518c-3p was enriched by AGO2 compared to IgG in A549 and H1299 cells; E. LINC00668 knockdown upregulated miR-518c-3p expression; F. miR-518c-3p inhibition decreased miR-518c-3p expression mediated by LINC00668 silencing. Data were analyzed by unpaired student’s 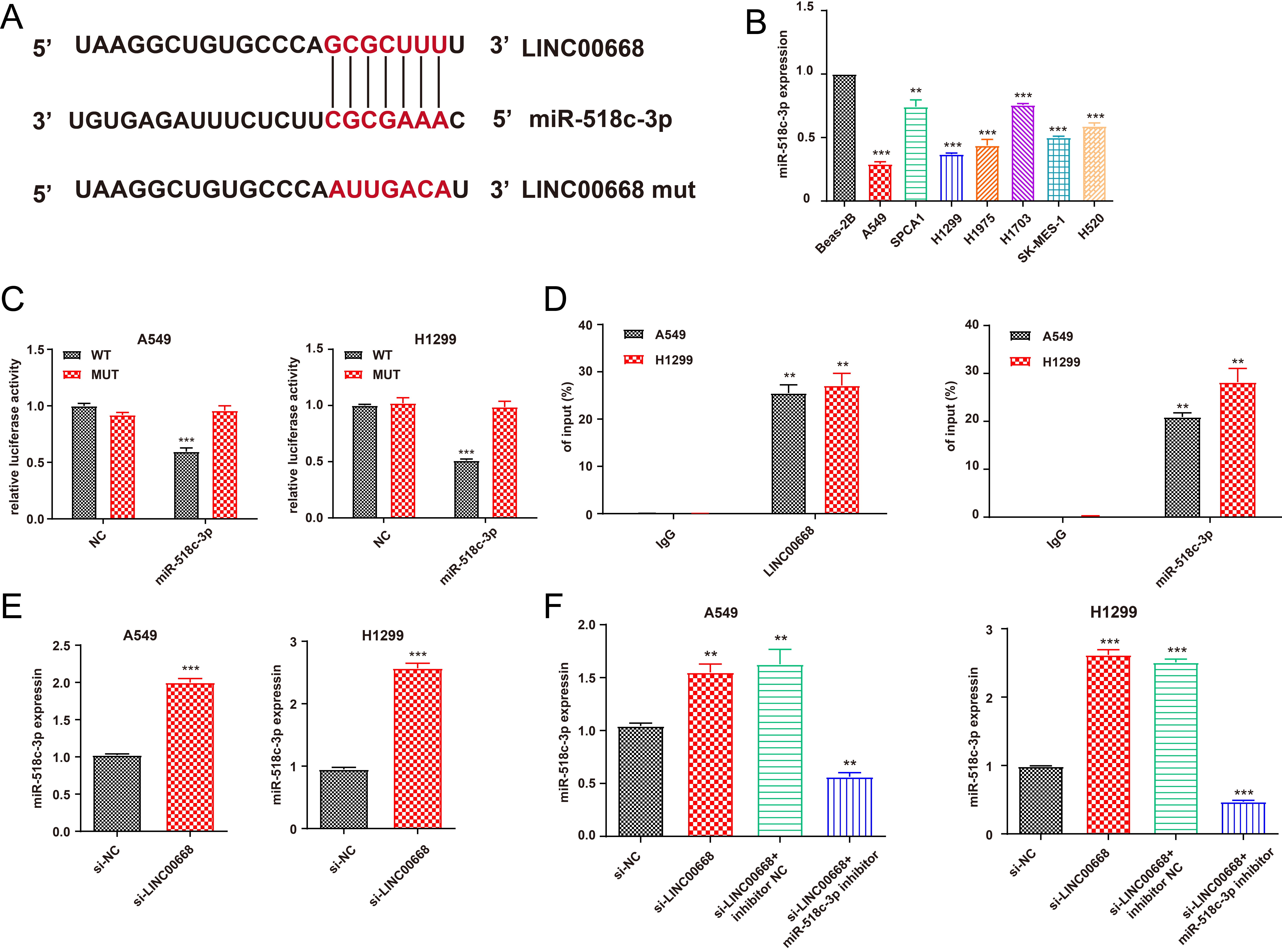
LncRNA-miRNA-mRNA was a crucial axis through which lncRNAs functioned in the carcinogenesis and progression of NSCLC. Using the ENCORI database, we identified miR-518c-3p as a highly potential miRNA to interact with LINC00668, and the binding site was illustrated in Fig. 2A. We further evaluated the expression of miR-518c-3p in several NSCLC cell lines and found that it was significantly downregulated in A549 and H1299 cells (Fig. 2B). Additionally, we constructed the LINC00668 mut plasmid with altered nucleotides at the binding sites for comparison, and the dual-luciferase reporter assays revealed that miR-518c-3p mimics weakened the luciferase activity in the WT group (Fig. 2C). Moreover, the RIP assays demonstrated that miR-518c-3p was significantly enriched by AGO2 compared to IgG, indicating its interaction with LINC00668 (Fig. 2D). Furthermore, LINC00668 knockdown significantly upregulated the expression of miR-518c-3p in A549 and H1299 cells (Fig. 2E). The qRT-PCR results further revealed that the miR-518c-3p inhibitor partially reversed the upregulation of miR-518c-3p induced by LINC00668 silencing (Fig. 2F).
miR-518c-3p inhibition rescued the decreased cell function mediated by LINC00668 knockdown. A–B. miR-518c-3p suppression partly retrieved the suppressed abilities of proliferation that LINC00668 knockdown mediated in NSCLC cell lines (A: CCK8; B: colony formation); C. miR-518c-3p inhibition decreased the elevated apoptotic cells mediated by LINC00668 knockdown in A549 and H1299 cells; D–E. miR-518c-3p partially rescued the inhibited abilities of invasion (D) and migration(E) induced by LINC00668 silencing. Data were analyzed by unpaired student’s 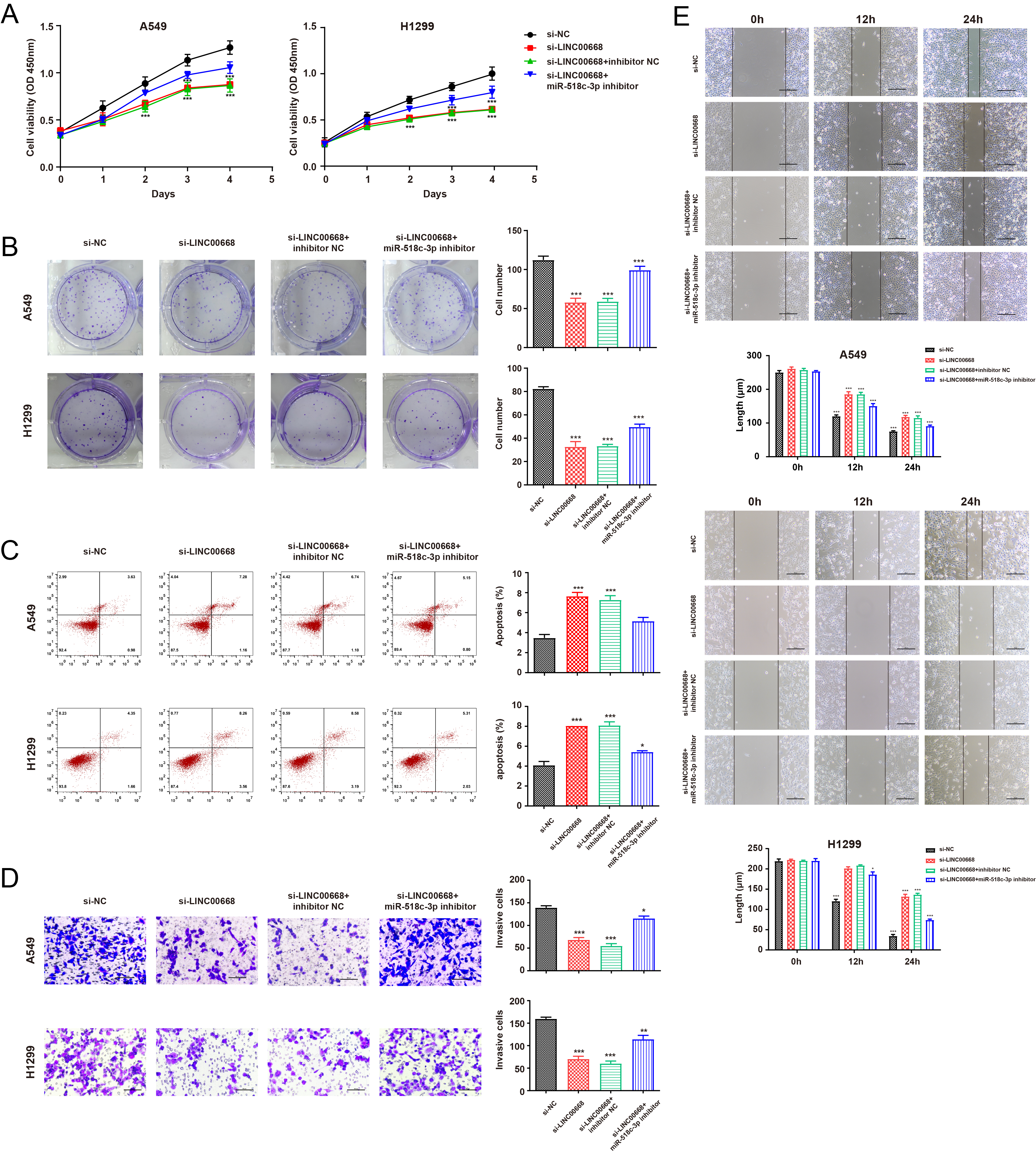
Subsequently, we conducted several rescue assays to investigate the biological function of miR-518c-3p in NSCLC cells. As shown in Fig. 3A and B, miR-518c-3p inhibition partially rescued the declined proliferation ability of NSCLC cells mediated by LINC00668 knockdown, as demonstrated by CCK8 and colony forming assays. Moreover, the inhibition of miR-518c-3p reversed the increased apoptosis rate caused by LINC00668 suppression (Fig. 3C). The wound healing and transwell assays further indicated that the repression of miR-518c-3p recovered cell migration and invasion, which had declined due to LINC00668 knockdown (Fig. 3D–F).
TRIP4 was upregulated in NSCLC tumor tissues and was negatively regulated by miR-518c-3p. A. miR-518c-3p had a binding site for TRIP4 basing on Starbase; B. Dual-Luciferase assays were utilized to detect the luciferase activities in A549 and H1299 cells after transfected with miRNAs (NC, miR-518-3p) and luciferase reporter vectors (WT and MUT) respectively; C. TRIP4 interacted with miR-518c-3p by RIP detection using AGO2 antibody in A549 and H1299; D–E. miR-518-3p negatively regulated TRIP4 at mRNA (D) and protein (E) levels; F. miR-518c-3p expression was negatively correlated to LINC00668 and TRIP4 expression using Pearson correlation analysis; G. TRIP4 was upregulated in tumor tissues compared to normal in 7 paired tissues. H. The IHC staining of TRIP4 in NSCLC tumors and normal tissues. Data were analyzed by unpaired student’s 
LINC00668 silencing downregulated TRIP4 and miR-518c-3p inhibition could partly rescue such decline. A. Suppression of miR-518c-3p partially rescued the downregulated TRIP4 expression mediated by LINC00668 silencing at mRNA level in NSCLC cells; B–C. Inhibition of miR-518c-3p partially retrieved the decreasing TRIP4 mediated by LINC00668 knockdown at protein level in A549 (B) and H1299 (C) cells. D–E. The transfection of pcDNA3.1-TRIP4 was estimated by RT-qPCR (D) and western blot assays (E). Data were analyzed by unpaired student’s 
TRIP4 rescued the impaired cell function mediated by LINC00668 downregulation in NSCLC cells. A–B. TRIP4 partly retrieved the suppressed abilities of proliferation that LINC00668 knockdown mediated in A549 and H1299 cells (A: CCK8; H: colony formation); C. TRIP4 overexpression decreased the elevated apoptotic cells mediated by the inhibition of LINC00668 in NSCLC cell lines; D–E. TRIP4 partially rescued the inhibited abilities of invasion (D) and migration(E) induced by LINC00668 knockdown. Data were analyzed by unpaired student’s 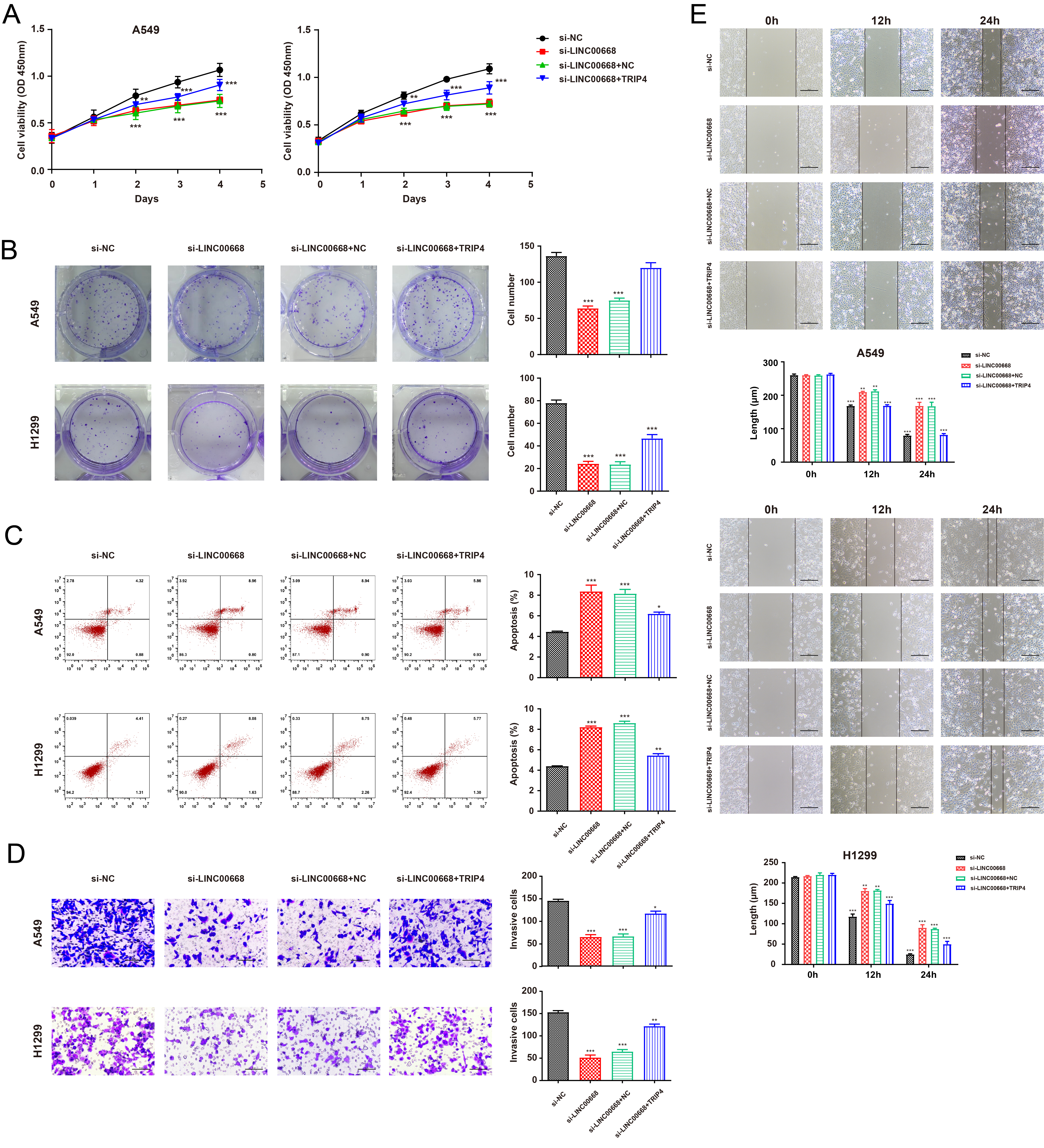
TRIP4 (Thyroid hormone receptor interactor 4, TRIP4) was predicted to interact with miR 518c-3p by the Starbase database and the potential binding site was illustrated in Fig. 4A. The dual-luciferase reporter assays demonstrated that miR-518c-3p mimics significantly decreased luciferase activity in both A549 and H1299 cells (Fig. 4B). To further validate the interaction between miR-518c-3p and TRIP4, RIP assays were performed, which revealed a significantly higher enrichment of TRIP4 and miR-518c-3p by AGO2 compared with IgG, as shown in Fig. 4C. The expression of TRIP4 was found to be downregulated at both the mRNA and protein levels after transfection with miR-518c-3p mimics, and conversely, upregulated after suppression of miR-518c-3p (Fig. 4D–E). Furthermore, miR-518c-3p expression was negatively correlated to both LINC00668 and TRIP4 expression (Fig. 4F). The expression of TRIP4 was then examined in 7 NSCLC tumor tissues and normal tissues using western blot assays. As Fig. 4G revealed, TRIP4 was significantly upregulated in NSCLC tumor tissues. The same result was observed in the IHC staining of TRIP4 (Fig. 4H).
The RT-qPCR demonstrated that LINC00668 silencing significantly downregulated the expression of TRIP4 at both the mRNA and protein levels, while inhibition of miR-518c-3p partly retrieved the decreased TRIP4 expression induced by LINC00668 knockdown, as shown in Fig. 5A–C. In light of TRIP4’s potential role as a downstream gene regulated by LINC00668, we synthesized the overexpression plasmid, pcDNA3.1-TRIP4. As evidenced by Fig. 5D–E, pcDNA3.1-TRIP4 effectively elevated TRIP4 expression at both mRNA and protein levels in A549 and H1299 cell lines. Functional assays including CCK8 and colony formation assays revealed that overexpression of TRIP4 countered the inhibitory effect of LINC00668 knockdown on NSCLC cell proliferation (Fig. 6A–B). Moreover, TRIP4 partially attenuated the elevated apoptosis rate induced by LINC00668 suppression (Fig. 6C). Additionally, upregulation of TRIP4 rescued the migration and invasion abilities of NSCLC cells that were reduced by LINC00668 knockdown (Fig. 6D–E). Collectively, these findings demonstrate that LINC00668 promoted NSCLC tumorigenesis and progression through modulation of TRIP4 expression.
Discussion
Lung cancer was the primary cause of cancer-related deaths and posed a significant threat to global health. Recent evidence has shown that long non-coding RNAs (lncRNAs) such as MALAT1 [18], CCAT2 [19], HOTAIR [20], and others play oncogenic or suppressive roles in NSCLC, which could have clinical significance for prognosis and treatment. Among these lncRNAs, LINC00668 has been identified to play oncogenic roles in the development of many malignancies, including breast cancer [15], gastric cancer [21], glioma [17], hepatocellular carcinoma [22], and colorectal cancer [23] as well as lung cancer. For instance, STAT3-induced LINC00668 was overexpressed in patients with NSCLC, and LINC00668/miR-193a/KLF7 pathway was identified [24]. Some compounds from traditional Chinese medicine played anticancer roles through LINC00668. Ophiopogonin-B (OP-B) inhibited metastasis of A549 cells via the LINC00668/miR-432-5p/EMT axis [25]. Sodium new houttuyfonate (SNH) was found to inhibit the metastasis of NSCLC cells. The underlying mechanism may associate with the LINC00668/miR-147a/Slug axis [26]. Consistent with the existing investigations, we found that LINC00668 was significantly upregulated in NSCLC tumor tissues and cell lines. Our in vitro experiments also revealed that LINC00668 silencing significantly inhibited NSCLC cell proliferation, invasion, and migration, while promoting cell apoptosis, indicating that LINC00668 acts as an oncogene in NSCLC development.
According to the competing endogenous RNA (ceRNA) theory, lncRNAs engaged in communication and co-regulation by competing for or interacting with common microRNAs, thus leading to the degradation and inhibition of downstream genes. These microRNAs are small non-coding RNAs that play crucial roles in post-transcriptional regulation. The ceRNA theory of lncRNAs has gained widespread acceptance among a growing body of research. For example, BC069792 sponged miR-658 and miR-4739 to upregulate KCNQ4, thus inhibiting JAK2 and p-AKT to suppress breast cancer progression [27]. LncRNA TINCR sponged miR-199a-5p to upregulate USP20 mRNA stability and therefore promote PD-L1 expression in breast cancer [28]. Ferroptosis-induced lncRNA NEAT1 promoted hepatocellular carcinoma proliferation through miR-362-3p/MIOX axis [29]. Given the crucial role of ceRNA mechanism in the progression of malignancies, we hypothesized that LINC00668 could facilitate NSCLC development by sponging miRNAs. We predicted the potential miRNAs that could bind to LINC00668 using the StarBase database and identified miR-518c-3p. Through a combination of RIP assays, RT-qPCR assays, and dual-luciferase reporter assays, it was demonstrated that LINC00668 exerted a negative regulatory effect on the expression of miR-518c-3p. MiR-518c-3p has been found to be dysregulated in various types of tumors and functions as a tumor suppressor [21, 30]. Our rescue experiments showed that miR-518c-3p suppression could reverse the inhibitory effects of LINC00668 inhibition on NSCLC progression, indicating that LINC00668 acts as a sponge for miR-518c-3p. MiRNAs primarily functioned by targeting mRNAs of downstream genes [31, 32]. We predicted the potential miR-518c-3p-targeted mRNAs through the StarBase database and identified TRIP4 mRNA as a potential binding site of miR-518c-3p. TRIP4 is located in 15q22.31 and has been shown to act as an oncogene in various malignancies. For instance, TRIP4 promoted glioma progression by activating DDIT4 and mTOR signaling [33]. TRIP4 activated MAPK, PI3K/AKT, and hTERT signaling to facilitate cervical cancer growth and metastasis [34]. Additionally, TRIP4 was targeted by miR-518-3p in colorectal cancer and the latter could inhibit tumor proliferation, invasion, and migration [35]. TRIP4 facilitated the growth of melanoma by influencing the expression of COX-2 and iNOS [36]. This effect was achieved in two ways: firstly, by indirectly activating NF-
In conclusion, our study has shed light on the significance of lncRNA LINC00668 in the development and progression of NSCLC. Mechanistically, LINC00668 sponged miR-518c-3p, leading to the upregulation of TRIP4, which in turn contributed to the progression of NSCLC, including cell proliferation, invasion, and migration. Moreover, the study highlighted the important roles of lncRNAs and miRNAs in cancer progression, suggesting that the ceRNA mechanism may be an essential process in the development of malignant tumors. Future studies should investigate the regulatory networks involving lncRNAs, miRNAs, and mRNAs to provide a more comprehensive understanding of the pathogenic mechanisms involved in NSCLC. Ultimately, the identification of LINC00668 in NSCLC may contribute to the development of diagnostic tests, personalized medicine, prognostic indicators, therapeutic targets, and preventive strategies. However, it is essential to conduct more rigorous studies, validate these findings in larger cohorts, and engage in collaborative efforts between researchers, clinicians, and industry partners to translate the discoveries of LINC00668 into practical clinical applications.
Footnotes
Acknowledgments
This work was supported by key projects of Science and Technology Development Fund of Pukou Branch of Jiangsu Province Hospital (KJ2021-8, KJ2022-7).
Conflict of interest
The authors declare that they have no conflict of interest.
Contributions
Conception: L. Chen.
Interpretation or analysis of data: Z.B. Lu, Z.C. Xiao, Q. Wang.
Preparation of the manuscript: Z.B. Lu, Z.C. Xiao, Q. Wang.
Revision for important intellectual content: C.F, Pan, Y. Xia, W.B. Wu.
Supervision: L. Chen.