
Abstract
BACKGROUND:
Haemochromatosis is an iron-storage disease with different genetic mutations, characterized by an increased intestinal absorption of iron, resulting in a deposition of excessive amounts of iron in parenchymal cells. When the iron is released in the blood, it is left in an unliganded form, where it can participate in Haber-Weiss and Fenton reactions, creating hydroxyl radicals. Erythrocytes (RBCs) are particularly vulnerable to hydroxyl radical damage, which can result in eryptosis (programmed cell death similar to apoptosis).
STUDY DESIGN AND METHODS:
Here, we used flow cytometry to study the presence of eryptosis in the main genotypic variations of HFE (heterozygous and homozygous C282Y; H63D; C282Y/H63D). We also viewed RBCs from the different mutations using super-resolution Airyscan confocal microscopy.
RESULTS:
Flow cytometry showed significant changes in membrane biochemistry, indicated by the presence of phosphatidylserine (PS) proteins on the outer leaflet of the membrane, as well as increased intracellular calpain. This was found in all of the studied mutations. Airyscan fluorescence revealed PS flip and also microparticles from RBCs. Such microparticles are known to be pro-inflammatory.
CONCLUSION:
We conclude that RBC pathology is present in all the studied HFE mutations, even in low penetrance mutations, and this might affect rheology in these individuals.
Introduction
Haemochromatosis (HFE-associated-hereditary hemochromatosis (HFE-HH)) is an iron-storage disease with different genetic mutations. It is characterized with an increased intestinal absorption of iron, resulting in a deposition of excessive amounts in parenchymal cells, due to the low production of the hormone hepcidin. This may eventually result in tissue damage and organ failure [1–3]. Classical HFE-HH is associated with mutations in the hereditary hemochromatosis gene (HFE); C282Y homozygotes or C282Y/H63D compound heterozygotes [4]. It is thought that C282Y homozygotes account for up to 90% of clinical cases of the condition, and that H63D and S65C homozygosity typically cause only mild disease or no clinical consequences [5].
One of the main medical complications of uncontrolled or excessive iron (of which serum ferritin (SF) levels are seen as a prominent indicator); is tissue damage derived from free radical toxicity [6–10]. HFE-HH is commonly (but not inevitably) associated with a persistent elevation of SF levels [11]. Elevated SF levels in turn, correlates with biomarkers of cell damage, in particular hydroxyl radical formation (and oxidative stress) and therefore, ultimately pathophysiology and disease [12, 13]. In a 2014 review [13] we suggested that “serum ferritin” presents a paradox, as the iron storage protein ferritin is not synthesized in serum, yet is to be found there, and we argued that SF arises from damaged cells, and is thus a marker of cellular damage. The current understanding of SF’s role as an intracellular iron storage protein, gives no explanation of why it should even exist in serum. We reasoned that SF leaks from damaged cells, losing most of its iron on the way, leaving it in an unliganded form, that can stimulate further cell damage [13, 14]. It can participate in Haber-Weiss and Fenton reactions, creating hydroxyl radicals. Hydroxyl radicals and an oxidative environment may lead to RBC cell death, or eryptosis.
We have shown that under laboratory conditions, in the presence of iron, healthy RBCs undergo a shape and membrane structural change and suggested that this was possibly due to the oxidative environment created by the presence of increased iron [15]. Also in individuals with HFE-HH and hyperferritinaemia individuals, RBCs have a changed morphology, where they are distorted and have a much greater axial ratio compared to the typical discoid appearance seen in healthy individuals [12]. In this 2014 study, a changed RBC morphology was found in HFE-HH individuals, with high, as well as normal SF levels. However, in the presence of iron chelators and free radical trapping agents, the shape of the RBCs reverted back to the typical discoid shape, suggesting that the aberrant morphology is caused, at least in part, by unliganded (‘free’) iron, that are released from the SF molecule. RBCs circulating in a high (unliganded) iron environment may therefore have a compromised physiological function. An important observation from this research was a changed RBC morphology (within even those individuals with the H63D (heterozygous) mutation, as was previously seen with scanning electron microscopy and light microscopy [12]. A parallel was made with sickle cell disease, where altered RBC morphology contributes to pathology, as the deformed RBCs struggle to pass through blood capillaries, often leading to stroke [16–19]. These shape changes could be associated with changes seen during eryptosis, which is the term used for programmed RBC death; this type of cell death happens in RBCs that do not have nuclei or mitochondria, and is comparable to apoptosis [20–23].
Eryptosis is characterized by cell shrinkage and cell membrane scrambling and is stimulated by calcium entry through Ca(2+)-permeable, PGE2-activated cation channels, by ceramide, caspases, calpain, complement, hyperosmotic shock, energy depletion, oxidative stress, and deranged activity of several kinases [24–30]. When Cl- is removed it causes the formation of PGE2 and COX activation, leading to arachidonic acid formation and PGE2 efflux, followed by stimulation and opening of the membrane Ca2+-channels. Oxidative stress may also cause the Ca2+ channel to open [31]. Osmotic stress can result in RBC shrinkage due to Ca2+ entry [32]. Increased cytosolic Ca2+ activates Ca2+-sensitive K+ channels, resulting in the subsequent exit of KCl and cell shrinkage [26]. Ca2+ entry also stimulates sphingomyelinase to form ceramide [32] and both Ca2+ and ceramide activates scramblase, resulting in phosphatidylserine (PS) exposure that represents a breakdown in asymmetry of the RBC cell membrane [26].
PS is usually found to the inner leaflet of the RBC and its externalisation requires both inhibition of the flippase and activation of the scramblase and both may follow from elevation of intracellular Ca2+ [33]. Annexin-V-binding to PS was determined by flow cytometry as a measure of PS exposure at the RBC surface. PS presence on the outer membrane is indicative of changes to the membrane of the RBC and together with an increase in calpain suggests the presence of eryptosis. If there is increased binding of Annexin-V to PS, some PS has externalized. It is usually most prominently present on the inside membrane envelope, but during eryptosis, it is found on the outer membrane [20–23]. COX mediates the formation of prostaglandins from arachidonate and its presence may suggest an increased prostanoid signalling; this in turn may show eryptosis. Anti-calpain S1 marker binds to circulating intracellular calpain 1 which is a Ca2+ -dependent cysteine protease, which binds to the Ca-channels on the RBC membrane. If binding by this marker is increased, it is an indication of an increased amount of calpain 1. This will result in an increased modelling of the cytoskeletal properties of RBC membranes. Elevated cytosolic free Ca2+ concentrations is known to activate μ-calpain, which degrades components of the cytoskeleton, like the ankyrin R-complex, and this also leads to membrane blebbing [34].
Oxidative stress have been demonstrated to damage RBCs from healthy donors by decreasing their deformability and aggregability, and by increasing their contribution to the processes of thrombus formation [36]. An increase in the number and size of erythrocyte aggregates relates to an increase in blood viscosity and hence, an increase in blood flow resistance – particularly in diseased vessels. This leads to restriction or even stagnation of blood flow [36, 37]. The purpose of erythrocyte deformability is to ensure efficient tissue perfusion. Reducing this capability does not only lead to a decrease in tissue oxygenation, but also shortens the lifetime of red blood cells [38]. Hemorheology can be influenced by blood viscosity, the deformability of erythrocytes (elongation index RBC) and aggregation (aggregation index RBC) [39]. Erythrocytes therefore plays an important role in hemorheology and should be monitored closely.
Here, we used flow cytometry to quantify the percentage RBCs with biochemical membrane changes in individuals with different HFE mutations, including C282Y (homozygous and heterozygous), H63D (homozygous and heterozygous) and C282Y/H63D. These individuals had both high and normal serum ferritin levels (at diagnoses). We compared the results to percentage change in RBCs from healthy individuals. We also visually analysed RBCs from HFE and healthy individuals using super-resolution Airyscan confocal microscopy.
Materials and methods
Patient and control demographics
Blood was collected from healthy volunteers and consenting patients with diagnosed different HFE mutations, including C282Y (homozygous and heterozygous), H63D (homozygous and heterozygous) and C282Y/H63D. HFE-HH blood was obtained from a routine genetics laboratory (AMPATH National Reference Laboratory) that had been sent there for genetic testing. Ethical approval for this study was granted at the University of Pretoria, South Africa, HUMAN ETHICS COMMITTEE: FACULTY OF HEALTH SCIENCES, under the name J N Du Plooy, 496/2015.
For control subjects, informed consent was obtained from healthy individuals who do not smoke and are not on any chronic medication, and with iron levels within the normal serum ferritin levels, as measured by a pathology laboratory (AMPATH South Africa) – for a detailed discussion on serum ferritin levels see [12]. All samples were tested within 72 hours from time of collection. (See supplementary data for demographics and serum ferritin levels of healthy individuals and those with a HFE mutation).
Flow cytometry
A number of antibodies are known markers for eryptosis, were used to examine RBCs (see Table 1).
Antibodies used in this study
Antibodies used in this study
After blood was collected, 1 ml was transferred to an eppindorf and centrifuged at 1000 rpm for two minutes at room temperature to isolate the RBCs. The supernatant was discarded (plasma, leukocytes and platelets) and the remaining RBC pellet was washed twice with a 0.15 M NaPO4 buffer for 3 minutes. In order to eliminate spectral overlap each antibody (see Table 1) was added separately.
For Annexin-V staining, 100μl of the washed RBCs were transferred to an eppindorf tube and then washed twice in Annexin-V binding buffer (BioLegend,422201) for 3 minutes, followed by re-suspending the RBCs in 100 ul of Annexin-V binding buffer and staining with 5μl Annexin-V (BioLegend,640906). Two gates, M1 and M2, were placed to set an arbitrary threshold between PS negative cells (PS-) and PS positive cells (PS+) respectively, the same was done for calpain.
COX-2 was included as an additional possible indicator of eryptosis. For anti-COX-2 and calpain staining the RBCs were permeabilized using 10μl of a 0.01% Triton X-100 solution for every 100μl of whole blood and incubated for 3 minutes. The samples were then washed 3 times with a 0.15 M NaPO4 buffer, ensuring all residual Triton X-100 was removed and subsequently stained with the respective antibodies. For the remaining antibodies, the washed RBCs were re-suspended in phosphate buffered saline (PBS) and 100μl of the suspended RBCs were transferred to eppindorf tubes for staining with 100μl 2 mM Fluo-3 AM (Abcam,ab145254), 5μl Anti-Cox-2 (Abcam,ab95838) and 0.5μl Anti-Calpain S1 (Abcam,ab92333). (Note that Calpain S1 is synonymous with μ-calpain [40]).
All samples were then incubated at room temperature in the dark for 1 hour, followed by a 2x-washing step (Annexin-V samples with binding buffer and the rest with PBS). The cells were then transferred to flow cytometry tubes, the Annexin-V samples were resuspended in 400μl Annexin-V binding buffer and the rest of the stained cells with 900μl sheath fluid (BeckmanCoulter). All samples were analysed with a flow cytometer (FC 500, Beckman Coulter).
Forward scatter and 90°-side scatter were displayed on logarithmic scales. The RBCs were gated according to the morphological characteristics of the RBCs. Fluorescence of the various antibodies was determined using excitation and emission values listed in Table 1.
Statistical analysis was done using the non-parametric Mann-Whitney test (significance was taken as p < 0.05), and the fluorescence signal was expressed as arithmetic means ± standard error mean (SEM) as previously described by [28].
Super-resolution Airyscan confocal microscopy
Results from flow cytometry showed that there were increased eryptotic signal in HFE RBCs, even in RBCs from the low penetrance H63D/heterozygous individuals. In order to visualise increased eryptotic signal, we used super-resolution confocal microscopy and Airyscan technology. RBCs from 12 HFE-HH individuals were compared to RBCs from 3 healthy individuals. Samples were prepared using the Annexin-V marker, and prepared as described for flow cytometry. After the last wash step the RBCs were resuspended in binding buffer and 10μl of the suspension was mounted onto a glass slide and covered with a coverslip. The sample was viewed with the Zeiss LSM 800 confocal scanning microscope in Airyscan mode. Airyscan is an array detector that draws on the fact that a fluorescence microscope will image a point-like source as an extended airy disk producing an image with increased signal-to-noise ratio and resolution. The Plan-Apochromat 63x/1.4 Oil DIC M27 and Alpha Plan-Apochromat 100x/1.46 Oil DIC M27 Elyra objectives were used.
To visualize the RBCs, 2 different lasers were used with different filters and beam splitter overlaying the respective images to show which RBCs have a PS flip present on the membrane. To view all cells present on the slide, the 405 laser was used to excite naturally occurring fluorescence found in RBCs, and a red colour was assigned to this fluorescent signal. In an unstained sample, the 405 laser was used together with the 465–505 Band pass (BP) and 525 long pass (LP) filters and the 488/405 beam splitters. These settings showed that all the RBCs present on the slide have auto-fluorescence, and this auto-fluorescence could therefore be used as a contrasting method against the Annexin-V binding, in the case where PS flip is present. To visualize Annexin-V binding the 488 laser was used with the 420–480/495–550 BP filters and the 488/405 beam splitters, and is shown as a green fluorescence, indicating the presence of a PS flip on the RBC membranes.
Results
In the present study we determined whether there is an increased presence of RBCs with eryptosis in individuals with heterozygous and homozygous mutations of the HFE gene. Statistical analysis of the all the HFE-HH individuals versus the control (Table 2) showed that there were significant differences in both Annexin-V-binding (p < 0.0001) and calpain presence (p = 0.0366), indicating an increased eryptotic potential in the HFE-HH population as a group; therefore throughout the different HFE mutations. For Annexin-V the p-value was calculated using the arithmetic means [28] and indicate the amount of PS binding per RBC, the percentage of PS+ cells are shown in the M2 gate where the control group had 2.8% PS+ cells and the HFE-HH group had 6.95% PS+ cells. Similarly, for calpain the p-value was calculated using the arithmetic means indicating the amount of intra-cellular calpain present in the RBCs. These results are shown in Table 2 as the median (95% Confidence interval; for the control group the AV median was 2.63 (2.29 to 2.89) and the HFE-HH group median was 4.9 (5.74 to 9.16). Regarding calpain the control group median was 1.68 (1.68 to 2.74) and the HFE-HH groups median was 1.9 (2.15 to 3.7) We note that these markers were also increased in the H63D-heterozygous individuals, who are not clinically diagnosed with haemochromatosis (reported as a silent phenotype).
Mann-Whitney results of the different markers of the HFE-HH individuals compared to healthy individuals
Mann-Whitney results of the different markers of the HFE-HH individuals compared to healthy individuals
Figure 1 shows a histogram and overlay of the pooled HFE-HH population versus the pooled healthy population for Annexin-V and anti-calpain S1 binding. The overlap in the control and HFE-HH anti-calpain S1 histograms may be attributed to the ubiquitous expression of calpain in humans [41–43]. One can however see a shift in the fluorescence intensity of the HFE-HH population to the right which was shown to be statistically significant using the Mann Whitney test.
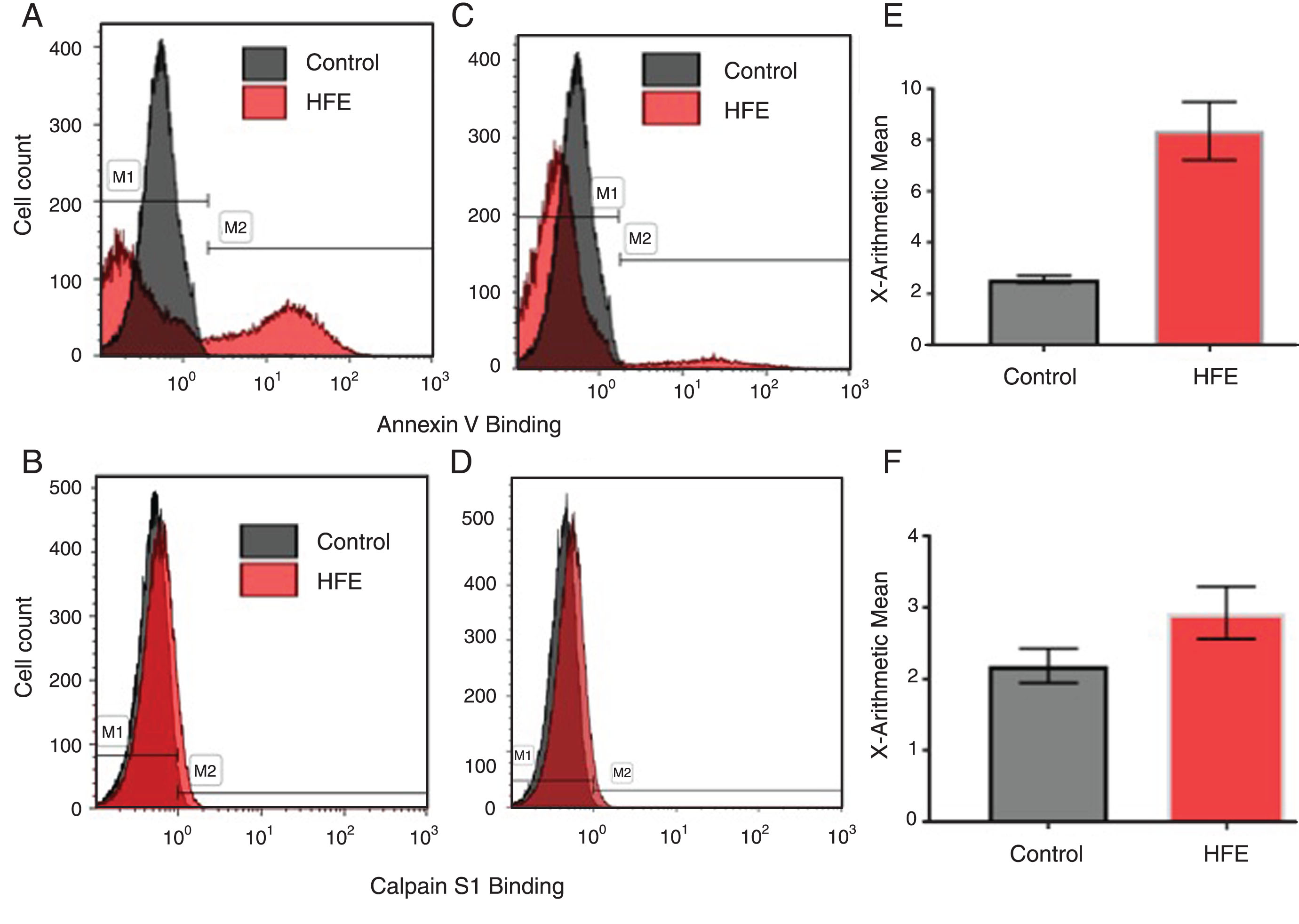
Overlay of healthy individuals, versus HFE genotypes for Annexin-V and anti-calpain S1 binding. A) Original histogram of Annexin-V binding of RBCs of one representative H63D homozygous HFE genotype (red area) and one representative healthy population (grey area) where gate M1 shows PS– cells and M2 shows PS+ cells. B) Original histogram of Calpain S1 binding of RBCs of one of the representative H63D homozygous HFE genotype (red area) and of one of the representative healthy individual (grey area). C) Original histogram of Annexin-V binding of RBCs of one representative H63D heterozygous HFE genotype (red area) and one representative healthy population (grey area) where gate M1 shows PS– cells and M2 shows PS+ cells. D) Original histogram of Calpain S1 binding of RBCs of one of the representative H63D heterozygous HFE genotype (red area) and of one of the representative healthy individual (grey area). E) Graph representing the arithmetic means of Annexin-V binding in the pooled HFE-HH group compared to the pooled healthy population±SEM. F) Graph representing the arithmetic means of Calpain S1 binding in the pooled HFE-HH group compared to the pooled healthy population±SEM.
Table 3 shows the results of each individual mutation relating to the two markers that were significantly different. The Annexin-V results are represented by the arithmetic means and percentage PS+ cells in the M2 gate while the Calpain results are represented by the arithmetic means. We also could not find any clear link with SF levels (as one might derive by the content of our supplementary data).
Annexin-V and Calpain results according to the HFE mutations
Following the significant differences present in Annexin-V binding between the healthy sample and the HFE-HH sample, we continued further to visually represent the flow cytometry results. To this end we prepared smears from healthy control samples and of another 12 HFE-HH individuals. Figure 2 shows how a typical healthy RBC sample looks like, while Fig. 3A to F show a collection of micrographs from a representative low phenotypic penetrance HFE-HH sample (H63D/+) (serum ferritin: 159 ng.mL-1). Figure 4 shows representative micrographs from C282Y/+, another H63D/+ and H63D/H63D. No or very little Annexin-V binding on the outer RBC membrane leaflet was observed in the healthy RBCs. This suggests that little PS-externalization happens in healthy cells. This confirms with results by various papers from the Lang research group, and others [20–23, 44–48]. RBCs of the H63D/heterozygous individual showed PS flip and RBC microparticle formation. RBC microparticles show both autofluorescence and Annexin-V binding areas.

A and B Micrographs from 2 different individuals, that is representative of a typical healthy RBC sample, stained with Annexin-V, showing no evidence of actual binding, due to the lack of a PS-flip. The visible red fluorescence is due to auto-fluorescence.
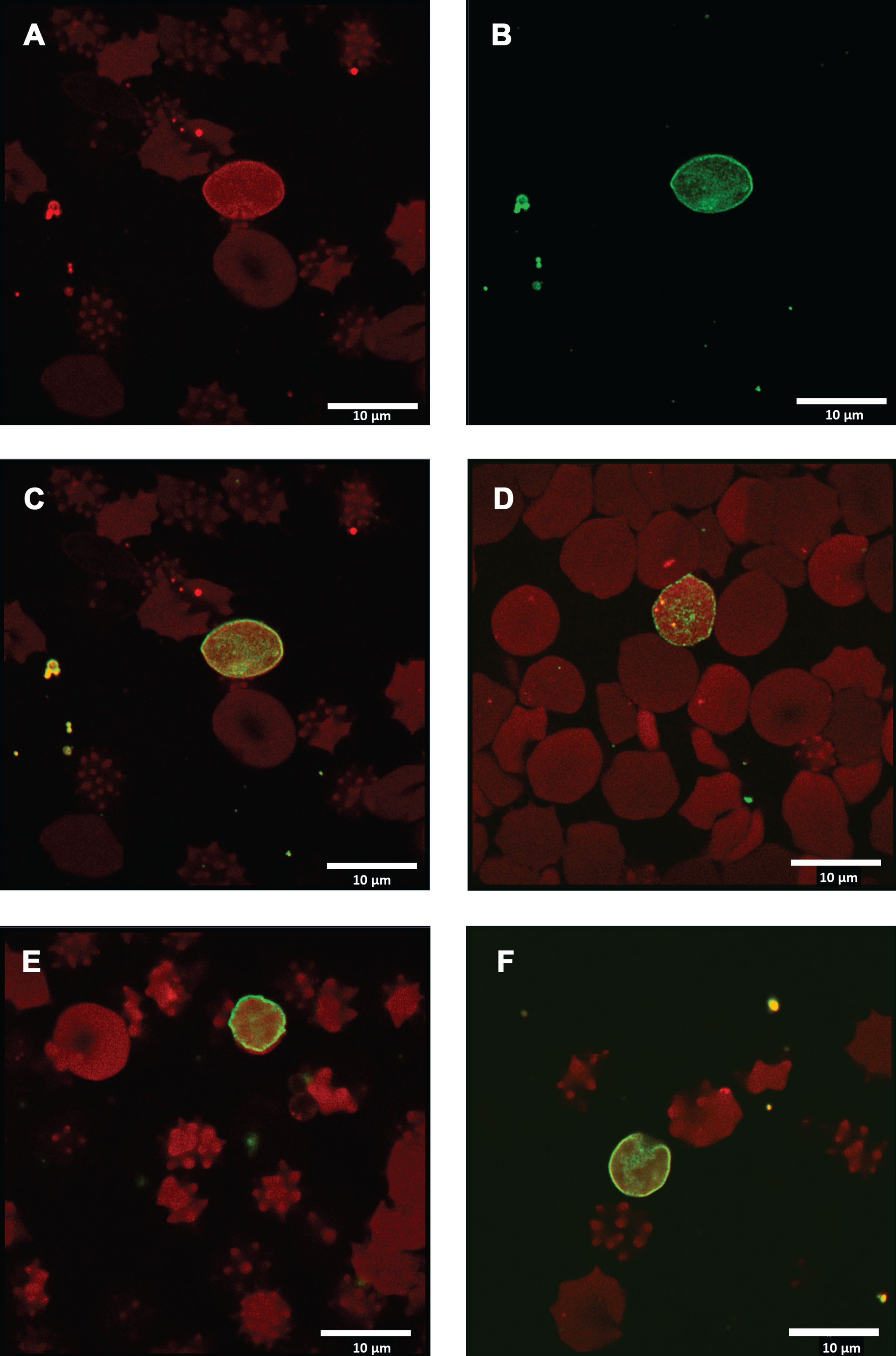
Micrographs from RBCs of a low penetrance H63D/heterozygous individual. A) Micrograph showing auto-fluorescence; B) Micrograph of same area, but with Annexin-V binding; C) Overlay of A and B to show both auto-fluorescence and Annexin-V binding. Arrows show microparticles from RBCs that also contain areas of PS flip. D, E and F) Additional micrographs showing both auto-fluorescence and Annexin-V binding.
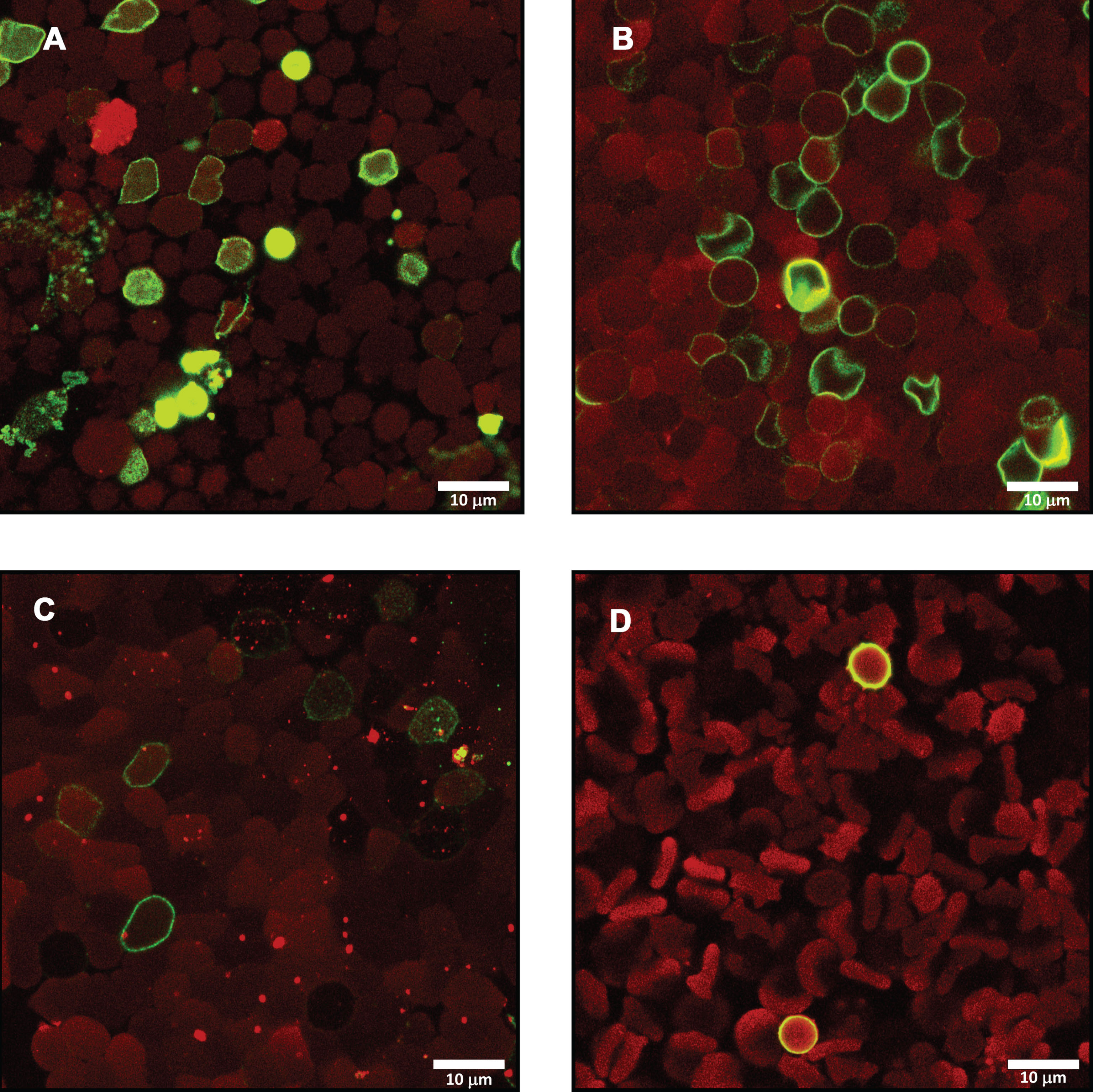
Micrographs showing RBCs from A) C282Y/+ (SF: 112 ng.mL-1).; B) H63D/+ (SF: 240 ng.mL-1).; C) H63D/H63D (SF: 218 ng.mL-1).; D) C282Y/C282Y (SF: 3129 ng.mL-1)., showing auto-fluorescence and Annexin-V binding.
Two important hallmarks of eryptosis are cell shrinkage and phospholipid scrambling of the cell membrane, where Ca2+ enters the cell and phosphatidylserine (PS) is translocated to the cell surface. Stimulators of Ca2+ entry into the RBC and eryptosis include oxidative stress, and an increase of intracellular Ca2+ leads to activation of Ca2+ sensitive K+ channels, resulting in cell shrinkage due to K+ exit, cell membrane hyperpolarization, Cl- exit and thus cellular loss of KCl with water [31].
Literature suggests that H63D heterozygosity is not associated with an increased risk for hemochromatosis, unless in compound heterozygosity with C282Y [1]. In a previous paper, morphological results suggested that, irrespective of the type of mutation that is present, HFE-HH RBCs have a changed RBC structure [12]. Here we show that in HFE RBCs with shape changes, there is PS externalization and this was confirmed with both flow cytometry and super-resolution confocal microscopy. This was also noted in RBCs from individuals with the H63D+ mutation, which have low phenotypic penetrance. Previous research by various groups that study eryptosis, also showed that PS externalization is associated with an increased presence of eryptosis [24, 46].
Although patients with H63D and S65C heterozygosity generally do not present with typical iron overload symptoms seen in classic HFE-HH patients, they may yet have an altered RBC morphology (eryptosis) which could have clinically significant implications for their health and could in the future be used as a potential health screen for these patients.
Our research group have previously shown that when increased SF levels are present in HFE individuals there is an accompanying change in RBC ultrastructure [12]. When FeCl3 is added to healthy whole blood morphological changes are also induced in RBCs, suggesting that unliganded iron itself causes morphological shape changes to RBCs [49]. We and others have shown that unliganded iron is an important cause oxidative damage through radical formation; and this damage was proposed to be related to the morphological changes in RBCs [13, 49]. In the present study, the RBCs of all the HFE-individuals showed eryptosis, even in individuals with normal to low SF levels. Ferritin has an H and L form that are structurally interchangeable and serum (L-)ferritin is usually measured with antibodies; only rarely is its iron content measured as well [13]. In our 2014 review [13] we argued that when SF measurements are done, SF is usually found to contain some iron, but nothing like its full complement. See papers on ferritin [50–56], suggesting that it has lost it, whether during or after effluxing from the cells in which it originates. We also suggested that: iron and SF can be regulated independently, excess SF protein is not of itself toxic in vivo, SF leaks from damaged cells, losing most of its iron on the way, and leaving that iron in an unliganded form that can impact negatively on health. This “dumped” unliganded iron can stimulate further cell damage [13].
Our results therefore support the possibility that unliganded iron in HFE-individuals participate in the Haber-Weiss and Fenton reactions, creating hydroxyl radicals which result in RBC cellular damage visible as eryptosis.
We also noted that in all mutations, there were membrane particles that showed both auto-fluorescence and Annexin-V binding. Microparticles are known to be pro-inflammatory. This is of great interest, as shedding of microparticles confirms RBC membrane blebbing, which is associated with eryptosis. These particles were not noted in flow cytometry, because they are simply too small to be detected using this technique. We provided autofluorescence as support that the particles come from RBCs as only RBC membrane particles will show autofluorescence in a whole blood sample consisting of platelets and a white blood cells. Calpain in particular, degrades the cytoskeleton, specifically the ankyrin R-complex, and this could lead to membrane blebbing and microparticle formation. A future study could investigate the presence of these microparticles further, by e.g. separating them from RBCs by low speed centrifugation to remove larger RBCs from the vesicle-enriched supernatant allowing better assessment as it might be an informative and potentially important cause of oxidative stress.
Our HFE-HH sample showed significant changes (p < 0.05) in the presence of both PS externalization and Calpain, suggesting that eryptosis is present in all individuals, regardless of the specific mutation or SF levels. Eryptosis is mostly a result of oxidative stress and/or hydroxyl radical formation. Multiple mechanisms have been identified whereby erythrocytes can add to the processes of pathological thrombus formation, and erythrocyte regulated hemorheology is the best known. Therefore the influence of erythrocytes integrity on hemorheology has an immense impact on hemostasis which can be studied by eryptosis. We conclude that RBC pathology is present in all the studied HFE mutations and its effects should be investigated further. A limitation of the study was that we did not have access to information about medication or whether the participants were smokers. This as well as the role of RBC microparticles could be addressed in more detail in a future study. More detailed analysis using e.g. western blot techniques could in future be done to further support our findings. RBC pathology, not previously recognized, might have far-reaching implications for the clinical understanding of this condition.
Compliance with ethical standards
The authors have nothing to disclose. Research involving Human Participants: HUMAN ETHICS COMMITTEE: FACULTY OF HEALTH SCIENCES (University of Pretoria), under the name J N Du Plooy, 496/2015.
Conflict of interest statement
There are no conflicts of interest to declare by any of the authors. EP, JB and JdP: paper concept and writing of paper. All authors approved of the submitted paper.