
Abstract
Chronic activation of coagulation is one of the features of sickle cell disease (SCD). Increased tissue factor expression, phosphatidylserine exposure, thrombin generation and fibrinolysis, as well as decreased levels of natural anticoagulants have been reported in SCD patients and in the mouse models of SCD. Consistent with this, patients with SCD are prone to develop thrombotic complications. In addition, the altered morphology of sickle red blood cells (RBC) may also alter the properties and dynamics of clot formation in SCD patients. Clinical data and results from animal models have revealed complex interactions between coagulation, chronic hemolysis, and inflammation suggesting that activation of coagulation may contribute to the pathophysiology of SCD.
Introduction
Sickle cell disease (SCD) is the most frequent genetic disorder in the world, and is caused by a single nucleotide mutation (adenine to thymine) in exon I of the β-globin gene that results in the substitution of glutamic acid for valine at the sixth amino acid position [1]. This mutation produces abnormal (sickle) hemoglobin (HbS) and homozygosity for the βS allele is the most common and severe form of SCD [2]. Under deoxygenated conditions, the hydrophobic residue of valine results in binding between hemoglobin tetramers and produces abnormal and large hemoglobin polymers that change normal biconcave red blood cells (RBCs) into less flexible, more fragile and irregularly shaped RBCs [1]. The pathophysiological complications of SCD are numerous, with hemolytic anemia and vaso-occlusive crises (VOC) being the primary clinical manifestations of the disease [3, 4]. Recently, several studies reported the activation of coagulation as a prominent feature of SCD [5–10] including a central role played by prothrombin in chronic inflammation and multiorgan complications in a transgenic sickle mouse model [11]. This so called “hypercoagulable state” observed in SCD is characterized by elevated levels of many biomarkers of thrombin generation [12, 13], enhanced platelet function [14], abnormal activation of the fibrinolytic system [12, 15], depletion of the natural anticoagulants [13], and increased tissue factor (TF) expression [8]. Furthermore, the enhanced externalization of phosphatidylserine (PS) on the outer membrane of sickle RBCs, particularly on membrane protrusions called “spicules,” enhances their prothrombotic phenotype [16]. Microparticles (MP) released by cells during apoptosis or activation, and hemolysis byproducts including free heme and MP are also known to activate coagulation via a TF-dependent pathway [8], PS exposure [17, 18] and the release of neutrophil extracellular traps (NETs) [19, 20].
Venous thromboembolism (VTE) has recently been recognized as a major cause of death in adults with SCD, affecting between 15 and 25% of SCD patients [21–23]. A number of factors contribute towards this increased risk for VTE in SCD, including platelet activation, enhanced fibrin deposition [24], and alterations in the RBC membrane [25]. Finally, the active role of RBCs in the clot contraction process has been recently demonstrated [26–28].
The objective of this review is to provide an update on the mechanisms leading to the activation of coagulation in SCD and highlight the contributions of this hypercoagulable state to the complex pathophysiology of SCD.
Coagulation cascade
The coagulation cascade consists of the extrinsic, intrinsic, and common pathways [29] (Fig. 1). The formation of the TF/FVIIa complex triggers coagulation by activating both FIX (a) and FX (b). Beyond its role in hemostasis, the TF/FVIIa complex contributes to multiple biological processes including inflammation [30]. The intrinsic pathway of coagulation is comprised of FXIIa, FXIa, FIXa and the cofactor FVIIIa (c). Activation of both pathways leads to thrombin generation through FXa-mediated activation of prothrombin (FII) in the presence of cofactor FVa (d). Following its initial generation, thrombin (FIIa) can also proteolytically activate FXI by a positive feedback loop (e) [31]. At the end of the coagulation cascade, thrombin (FIIa) cleaves fibrinogen into insoluble fibrin (f) [32]. The fibrin network is then stabilized by FXIIIa-mediated crosslinking of γ- and α- chains of fibrin (g) [33]. Clot lysis is mediated by plasmin, the first lysis factor, which is released from its zymogen plasminogen by serine proteases (tissue or urokinase plasminogen activator) (h) [32, 34].

Coagulation cascade: extrinsic, intrinsic and common pathways. TF, tissue factor; tPA, tissue plasminogen activator.
Higher thrombin generation
Elevated plasma levels of markers of thrombin generation such as thrombin anti-thrombin complexes (TAT) and prothrombin fragment 1.2 (F1.2) have been reported in adults [10, 35] and children with SCD [36–38]. Similarly, Chantrathammachart et al [6] reported elevated TAT levels in BERK and Townes mice, the most commonly used animal models of SCD [39, 40]. Additionally, mice treated with the anti-mouse TF antibody 1H1 had lower plasma TAT levels, suggesting that the increased thrombin generation observed in mouse models of SCD is TF-dependent [6]. Multiple studies have investigated the effect of SCD on thrombin generation potential in platelet poor plasma with conflicting results [41]. In children with SCD, Noubouossie et al [36] found significantly higher indices of thrombin generation in platelet poor plasma compared to controls. Whelihan et al [10] compared the effect of SCD on thrombin generation in platelet poor plasma and whole blood. They reported that thrombin generation was higher in whole blood and lower in plasma of SCD patients compared to healthy subjects. The lack of standardization of the thrombin generation assay may be one explanation for the inconsistent results between studies [10]. Moreover, the absence of RBCs and monocytes may also explain these discrepancies, as these cell populations demonstrate procoagulant activity in SCD [42].
Increased TF expression
TF is a 47-kDa transmembrane glycoprotein that initiates the extrinsic pathway of blood coagulation [43, 44]. Under physiological conditions, TF is constitutively expressed by adventitial cells within the vessel wall and is physically separated from its ligand FVII/FVIIa by an intact layer of endothelial cells [45]. In contrast, under pathological conditions including SCD, TF expression is induced on leukocytes [46–48] and endothelial cells [47, 49]. Key et al [42] reported higher levels of whole blood TF activity in patients with SCD, which correlated with TF-positive monocytes, TAT, and D-dimer [47]. During VOC, several studies have reported increased monocyte TF expression compared to steady state [46, 49], although others did not observe significant differences [42].
Solovey and colleagues reported that sickle mice with the most severe phenotype expressed the highest levels of endothelial cell TF in the lungs [48]. Further, hypoxia/reoxygenation conditions have also been shown to increase TF expression in pulmonary vein endothelium and monocytes in sickle mice with a less severe phenotype. Interestingly, Sparkenbaugh et al [50] demonstrated that low TF expression in all non-hematopoietic cells had no effect on plasma TAT levels in sickle mice suggesting that this cellular source of TF is not essential for thrombin generation. However, decreased TF expression in non-hematopoietic cells was associated with decreased plasma levels of interleukin-6 (IL-6) and slightly decreased lung myeloperoxidase (MPO) levels in sickle mice. In accordance with these results, Chantrathammachart et al [6] demonstrated that endothelial cell specific deletion of TF had no effect on plasma TAT levels, while it decreased the plasma concentration of IL-6. Thus, endothelial cell-derived TF appears to be involved in inflammation rather than activation of coagulation in transgenic sickle mice.
Pretreatment with lovastatin, which induces nitric oxide synthase and resultant NO production [51], suppressed the hypoxia/reoxygenation-induced endothelial TF expression in the lungs of sickle mice [48]. Unlike lovastatin, a pilot study with a short-term use of simvastatin in SCD resulted in a mild but insignificant decrease of plasma TF levels [52]. Furthermore, endothelial TF expression in sickle transgenic mice is regulated by endothelial nitric oxide synthase [53] and NF-KB (p50) activation in blood mononuclear cells but not endothelial cells [54].
Externalization of phosphatidylserine (PS)
PS is normally confined on the inner leaflet of the cell membrane but switches to the surface of the outer cell membrane in senescent RBCs, hence serving as an “eat me” signal [55, 56]. Flippase inactivation and scramblase activation via the cleavage of these two membrane proteins by caspases leads to PS exposure and disrupted asymmetrical phospholipid distribution in the plasma membrane [56]. PS can be exposed on the surface of mature sickle RBCs [57, 58], on the surface of reticulocytes [59] and on the surface of sickle RBC-derived microvesicles [60]. Increased PS exposure has been shown to correlate with the degree of dehydration in sickle RBCs [59, 61]. PS exposure can also be triggered by “apoptosis-like cell death” of erythrocytes, also referred to as eryptosis [7, 62].
Fluorescein isothiocyanate (FITC) labelled annexin V binds with strong affinity to PS and is typically used in conjunction with flow cytometry to analyze PS exposure [7, 64]. Significantly higher levels of annexin V labelling was observed on the surface of RBCs from SCD patients compared to RBCs from control subjects suggesting an increased loss of phospholipid asymmetry in these patients [63, 64]. Recently, Whelihan et al [10] estimated RBCs-PS exposure using FITC-labeled bovine lactadherin and reported results similar to annexin V.
PS exposure on the cell surface enhances the procoagulant activity [65] and the adhesive properties of sickle RBCs [7, 66]. Increased exposure of negatively charged PS acts as a binding site for enzymatic complexes, such as the serine proteases involved in coagulation. PS can also be recognized by macrophages, which consequently degrade the RBCs and help with their clearance [67]. SCD patients also have high levels of circulating PS-positive erythrocytes due to the impairment of spleen function [68, 69]. Thus, PS externalization plays a key role in the occurrence of thrombotic events [63, 64].
Surprisingly, Whelihan et al [10] did not find a significant correlation between RBC-PS externalization and circulating baseline TAT levels in SCD patients, which is in contrast with previous studies [16, 70, 71]. Also, in vitro data reported a modest negative correlation between the maximum rate of αTAT formation, the maximum TAT formed after extrinsic activation in SCD, and RBC-PS exposure. In addition, Whelihan et al [10] showed an inverse correlation between whole blood thrombin generation and RBC-PS exposure. These results may be explained by the fact that the maximum rates of thrombin generation were reported for an optimum of phospholipid concentrations between 25 and 100 μM [72] and above these levels, thrombin generation might be decreased. Other coagulation markers reported to correlate with PS positive erythrocytes include F1+2, D-dimers, and plasmin-antiplasmin complexes [13, 73].
Platelet activation
Increased platelet activation is another known factor observed in SCD patients at steady state [74–77], with further increases during VOC [78, 79], that could contribute to increased thrombotic complications [5]. Several mechanisms have been identified to explain platelet activation in SCD including hyposplenism [75] and hemolysis [80]. In healthy subjects, Helms et al [81] found that in vivo infusions of cell-free hemoglobin and ADP, released during hemolysis, enhanced platelet activation by decreasing NO bioavailability. Consistent with this, flow cytometry analysis of sickle blood showed a positive correlation between markers of platelet activation and hemolysis, and the in vitro effects of cell free hemoglobin on platelet activation was blocked by NO [80]. Further, supplementation with nitrate attenuated the platelet activation and adhesion to endothelial cells in a mouse model of hemolysis-induced inflammation and in sickle mice via a NO-dependent mechanism [82].
Experiments with healthy blood donors have showed that activated platelets express CD40 ligand (CD40L), a platelet-associated pro-inflammatory molecule [76]. Activated platelets can also induce TF expression on monocytes in a P-selectin dependent manner [83]. More than 30-fold increase in soluble CD40L was reported in SCD patients compared to healthy control subjects [76, 84]. In addition, a positive correlation between soluble CD40L (sCD40L) and plasma TF levels was observed, suggesting that CD40L may contribute to TF-dependent activation of coagulation in SCD patients [76]. Investigating the association between higher plasma concentrations of sCD40L and clinical manifestations revealed that SCD patients with sCD40L >112.9 pg/ml were at higher risk of acute chest syndrome than those with sCD40L <112.9 pg/ml [84]. The pro-thrombotic and pro-inflammatory marker tumor necrosis factor (TNF)-superfamily cytokine (TNFSF14) has also been shown to correlate with the activation of platelets in SCD. Platelets may be the main source of TNFSF14 in SCD patients [74].
Evidence of platelet activation in SCD has led to several clinical trials evaluating the effect of anti-platelet agents as therapeutics in SCD [85–88]. Eptifibatide, an inhibitor of αIIbβ3-mediated platelet adhesion to fibrinogen [89], was reported to decrease sCD40L levels and to blunt platelet aggregation in SCD patients in a phase 1 study [90]. Eptifibatide treatment appeared to be safe, and induced neither major bleeding events nor thrombocytopenia [91]. However, this treatment did not significantly reduce time to crisis resolution, hospital discharge, or opioid use. Another, antiplatelet drug Prasugrel, an oral thienopyridine drug, is particularly relevant in SCD since it can inhibit the ADP-dependent activation and aggregation of platelets [92]. Prasugrel was well-tolerated in children [93] and adults [85] with SCD. In a multicenter phase 2 study, 30 days of treatment with Prasugrel reduced both soluble P-selectin and P-selectin on the surface of platelets in SCD patients compared to control subjects. However, Prasugrel had only a moderate inhibitory effect on mean pain rate (percentage of days with pain) and intensity compared with placebo and did not reach statistical significance [94]. Recently, Heeney et al [95] evaluated the effects of Prasugrel in a phase 3 double-blind, placebo-controlled multinational study. They did not find any significant difference with respect to the rate of VOC events and related complications, or the rate of hospitalizations in SCD patients treated with Prasugrel in this study.
Depletion of natural anticoagulant
Protein C and protein S are vitamin K-dependent proteins with anticoagulant functions that are usually depleted in SCD patients either due to their decreased production or increased consumption. Overall, several human studies found lower protein C and protein S levels in adults [10, 96] and children with SCD [37, 97–100]. Furthermore, Tan et al [100] reported greater reduction in protein S and protein C levels in sickle cell children with a history of stroke versus children with SCD and absence of neurological sequelae. Chronic transfusion did not seem to reverse the depletion of these natural anticoagulants [98].
The activation of protein C is triggered by binding to the endothelial protein C receptor (EPCR) through thrombomodulin-bound thrombin. Activated protein C can dissociate from EPCR and acts as an antithrombotic protease by inactivating clotting factor Va (FVa) and FVIIIa [101]. It was demonstrated in vitro that RBCs from sickle cell patients provide a catalytic surface for FVa inactivation by activated protein C [102]. Consequently, chronic/persistent activation of coagulation in SCD may enhance the consumption of protein C. Indeed, the addition of thrombomodulin into the platelet-poor plasma enhanced endogenous thrombin potential, peak height, and velocity in children with SCD compared to race-matched controls [37]. In addition, murine protein C and arteriolar EPCR expression were found to be significantly reduced in the cerebral vasculature of sickle mice. Importantly, administration of activated protein C prevented the enhanced thrombosis observed in both the arterioles and venules of sickle mice [103].
The anticoagulant action of activated protein C is enhanced by several cofactors, such as protein S and PS [104]. Protein S had been shown to bind with high affinity to vesicles and dense irreversibly sickled RBCs in a calcium-dependent manner [105]. In SCD patients, Whelihan et al [10] found that lower protein S levels were inversely correlated with RBC-PS exposure, and confirmed the presence of protein S on the surface of sickle RBCs by flow cytometry. Thus, as previously suggested [13], lower protein S in SCD patients may result from higher clearance due to protein S binding to exposed PS on sickle RBCs. It has also been suggested that elevated FVIII levels in SCD patients may be an additional factor contributing to the depletion of protein C and protein S [10, 107].
Abnormal activation of the fibrinolytic system
SCD patients also demonstrate higher levels of fibrinolytic markers, such as D-dimers, fibrinopeptide A, and plasmin-antiplasmin complexes compared with race-matched controls [5, 109]. Compared to the values obtained during the admission for VOC, plasma levels of D-dimers are significantly elevated in children with SCD 48 hours later [37]. Consistent with these results, Tomer et al [12] found an association between SCD pain and elevated plasma levels of plasmin-antiplasmin and D-dimers. In addition, elevated plasma fibrinolytic activity measured by enzyme immunoassay was positively correlated with high frequency of pain episodes and negatively correlated with the time to the next painful episode [12]. In children with SCD, TF expression on monocytes was found to positively correlate with D-dimer and fibrinogen levels [46]. D-dimer levels were also reported to correlate positively with stroke history [5] and increased risk of cerebral silent infarcts, a severe and frequent complication in SCD children [108]. Recently, one year of HU treatment attenuated plasma levels of TAT and D-dimers in SCD patients, which was associated with a reduction in hemolysis markers and levels of cell adhesion molecules [5].
Hypercoagulability as part of the vicious cycle of sickle cell disease
Due to repeated polymerization and depolymerization cycles of HbS, RBCs eventually become irreversibly sickled, and consequently more adherent to the vascular endothelium and other vascular cells [110, 111]. Repeated sickling of RBCs results in premature senescence of one-third of these cells [53]. Sickle RBCs are less flexible, and more prone to aggregation and hemolysis than healthy RBCs [112, 113]. Major consequences of this are chronic hemolytic anemia resulting in increased levels of free heme [114] and VOC leading to painful crisis and ischemia/reperfusion injury in multiple organs [115]. These two major complications enhance vascular inflammation and oxidative stress, which further perpetuates the vicious cycle of SCD pathology (Fig. 2) [116]. Investigations into the role of coagulation in the pathophysiology of SCD [5, 18] have revealed a “crosstalk” between vascular inflammation and coagulation [18]. For instance, markers of thrombin generation were found to correlate with soluble vascular adhesion molecule-1 (sVCAM-1) in patients with SCD [5]. Moreover, Chantrathammachart et al [6] demonstrated that blocking TF not only reduced activation of coagulation but also attenuated inflammation and endothelial cell activation in mouse models of SCD. In the same way, low TF expression in sickle mice led to a drop in vascular congestion in the liver suggesting that TF plays a key role in vascular inflammation and cellular stasis [117]. One putative pathway that could mediate the regulation of inflammation via the TF/FVIIa complex may involve activation of protease-activated receptor-2 (PAR2) [118, 119]. Interestingly, both pharmacological inhibition of FXa or PAR-2 deficiency in non-hematopoietic cells attenuated systemic inflammation in sickle mice. Thus, PAR-2 could be a potential target to decrease vascular inflammation associated with SCD [120].
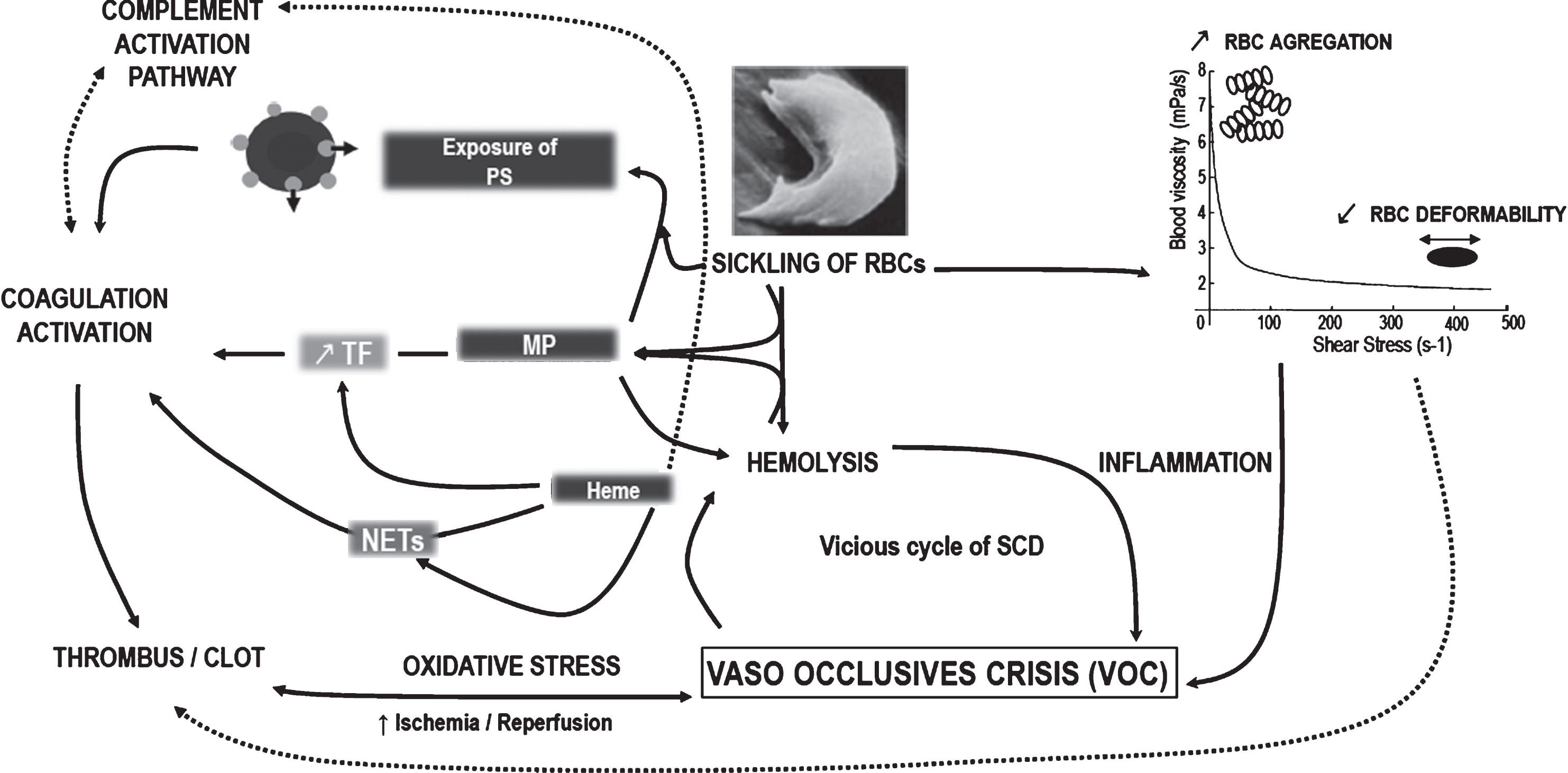
Activation of coagulation, part of the vicious cycle of sickle cell disease. Sickling of red blood cells (RBCs) led to hemorheological alterations including elevated strength of RBCs aggregates and decreased RBCs deformability that may affect clot formation [112, 113]. Also, sickle RBCs are more fragile and prone to hemolysis and to membrane alterations with phosphatidylserine (PS) exposure at their surface [55]. Thus, specific properties of sickle RBCs and byproducts released during chronic hemolysis induce inflammation and enhance the risk to trigger painful VOC at various organ levels [114]. The intermittent cessation of blood flow and consequent ischemia/reperfusion events may disturb the redox balance which in turn amplify hemolytic rate [115] and may increase thrombus/clot formation [145]. Free heme upregulates tissue factor (TF) expression on endothelial cells and white blood cells triggering the extrinsic pathway of coagulation [8, 135]. Free heme may also act as a convertase activating the complement system and increasing the coagulation. In the other way, heme-laden microparticles (MP) are able to transfer heme to vascular compartment and mediate inflammation, oxidative stress and VOC [140]. Heme can also activate coagulation via neutrophil extracellular traps (NETs) released by neutrophil upon activation. The pathway involved in the regulation of heme induced NETs formation could be oxidative stress-dependent [19]. Both TF [141, 149] and phosphatidylserine (PS)-exposed on MPs [36, 179] increase the procoagulant activity in SCD. Thus, by increasing TF exposure [42, 46–49], PS externalization [63–65] and NETs released [20, 138], the sickling of RBCs and the associated chronic hemolysis, may enhance the activation of coagulation. Increasing evidences reported that the activation of coagulation resulting from all those pathways not only induced thrombotic complications but also contribute to vascular inflammation that consequently exacerbate the vicious cycle of sickle cell disease (SCD). Dotted lines indicate the putative link in SCD patients, while complete arrows indicate the known relations in the pathophysiology of SCD.
Haptoglobin and hemopexin are the natural scavengers of hemoglobin and heme, respectively. Chronic hemolysis associated with SCD results in the depletion of both proteins and leads to increased plasma levels of cell free hemoglobin and heme. The excess of free heme is highly cytotoxic, exacerbates inflammation and oxidative stress, and enhances risk for VOC in SCD patients [121–123] and in transgenic sickle mice [124–126]. In 2012, Setty et al [47] observed that hemolysis might be a key factor contributing to the procoagulant phenotype in SCD. Below, we will discuss several mechanisms by which hemolysis can contribute to the procoagulant state in SCD [8, 127–129].
Heme promotes procoagulant activity
TF-dependent process
As previously mentioned, TF is the primary initiator of the extrinsic pathway of coagulation. Genetic reduction in TF expression or inhibition of TF activity attenuated plasma TAT levels in a mouse model of heme overload while inhibition of FXI did not affect heme-induced thrombin generation in this model. Thus, heme-induced activation of coagulation involves activation of the extrinsic but not the intrinsic pathway [8].
Treatment of endothelial cells with heme in vitro increases TF mRNA expression and activity in a concentration-dependent manner [128, 129]. The inhibition of NF-KB activation with sulfasalazine and curcumin inhibited heme-mediated TF mRNA expression in endothelial cells suggesting involvement of NF-KB in this process [129]. Furthermore, Rehani et al [128] found that heme also induced TF expression in blood monocytes. In vivo studies have partially confirmed these results, since TF-positive leukocytes were isolated from heme-treated mice whereas TF-positive endothelial cells were not observed in the mouse lung vasculature after heme treatment [8]. This suggests that the expression of endothelial cell TF observed in sickle mice is not simply mediated by increased levels of free heme [49]. Additionally, the treatment of sickle mice with recombinant hemopexin, which was previously shown to have beneficial effects on endothelial activation, oxidative stress, and VOC in SCD mice [124, 130], only partially blunted the activation of coagulation [8].
Recently, by investigating the dynamics of clot formation using a rotational thromboelastometry (ROTEM) experiment, De Souza et al [131] demonstrated that heme-mediated TF expression affected ex vivo coagulation activation of healthy whole blood samples. More specifically, the TF-dependent effects of heme could be involved during the initial steps of clot formation. It could be interesting to perform this assay with sickle blood since several studies have started to report alteration of the clot retraction process in SCD patients (paragraph “Alteration of clot formation in SCD”) but the underlying mechanisms remain unknown [27, 28].
Also, in a recent review describing the close interactions between the coagulation factors and complement system, SCD patients demonstrated increased levels of C5-b9 compared to healthy subjects [132]. Heme has been reported to activate the complement system including C5-b9, a membrane attack complex, which can render RBCs more prone to lysis [133] and exacerbate coagulation activation. Thus, we can hypothesize a putative role of a heme-associated imbalance of the complement system in the activation of coagulation in SCD.
NETs released
NETs are decondensed chromatin comprising extracellular DNA, nucleosomes, histones, and neutrophil proteases. NETs are released by neutrophils upon activation [134, 135] and are a double-edged sword with a protective role against invading pathogens [134] and a highly cytotoxic role during sterile inflammation [136]. In the past decade, NETs have also been reported to have procoagulant activity and contribute to venous thrombosis [137, 138]. In sickle patients, markers of NET formation (circulating nucleosomes and neutrophil activation) are increased at steady state and are further elevated during painful crisis [139]. Chen et al [19] identified heme as a potent regulator of neutrophil NETs release in sickle mouse model of VOC induced by TNF-α and in vitro. The underlying mechanism that could explain heme-induced NET formation could be oxidative stress-dependent. Importantly, hemopexin infusion prevented NETs formation in SCD mice and protected against TNFα-induced pathological complications. [19].
Microparticles (MPs) promote procoagulant activity
Chronic hemolysis in SCD patients can also trigger production of RBC-derived MPs [139–143]. Several studies found positive correlations between RBC-MPs and hemolytic components known to have strong relationships with markers of intravascular hemolysis in SCD patients [35, 144].
The role of MPs in SCD has recently been reviewed in detail [127] (see review from Romana et al in this issue), which highlight their notable role as biomarkers in coagulation activation. MPs can be defined as sub-micron vesicles derived from the plasma membrane of different cells including RBCs, platelets, monocytes, and endothelial cells [127]. Levels of MPs can reach a 5- to 6-fold increase in SCD patients versus healthy subjects at steady state [140, 141], and those levels can be further increased during painful VOC [141–143]. MPs derived from RBCs were reported to be the most prevalent in SCD [140, 145–148].
Several markers of coagulation activation like D-dimer, TAT, and F1+2, were also found to correlate positively with MPs numbers, including erythrocyte-derived MPs [35] and total-MPs [142]. Both TF and PS-exposed on MPs increase the procoagulant activity of MPs in SCD.
TF has been observed on MPs derived from both endothelial cells and monocytes in SCD patients [141, 149]. However, in contrast to that, van Beers and colleagues reported that the increased procoagulant activity in the plasma of sickle cell patients is associated with RBCs-derived MPs and is mediated by FXI- but not FVII-dependent mechanism [35]. Furthermore, Chantrathammachart et al [6] did not report a significant increase in MP-TF activity in transgenic sickle mouse models.
Meanwhile, MPs positive for PS [143], including RBCs and platelets-derived MPs [142], have also been shown to participate in the prothrombotic state in SCD. Also, circulating MPs [36] and RBC-derived MPs [150] with PS exposure at their surface explained the higher level of thrombin generation observed in SCD patients. Those authors further reported that HU treatment decreased thrombin generation by blunting RBCs-derived MPs and RBCs-MPs expressing PS+ (RBCs-MPs/PS+). It is necessary to point out that a recent study [151] reported no changes in RBCs-MPs concentration after 2 years of HU treatment in SCD patients. However, they observed a decrease in the density of PS on the RBCs-MPs suggesting that procoagulant RBCs-MPs/PS+ may explain hypercoagulable state in SCD patients. Thus MPs could be a highly potent biomarker of thrombin generation in SCD based on their levels and/or subtypes [127].
Alteration of clot formation in SCD
Recent data indicate that RBCs are not simply trapped within fibrin-rich thrombi but in contrast may actively affect the clot formation process [24, 26–28]. Cines et al [26] reported that RBCs are prone to change their shape from biconcave to polyhedral structures due to forces generated by platelets and the fibrin network in human coronary artery thrombi and in blood clot formed ex vivo. However, in SCD patients, clot contraction mediated by erythrocyte packing has been shown to be significantly altered [27]. Human sickle RBCs entrapped in the core of the clots (formed ex vivo) had amorphous shape, elongated structures, and higher fibrin content, with much more heterogeneity in deoxygenated conditions. Tutwiler et al [28] went further in the understanding of the role of RBCs into the clot contraction process. They described clot contraction as a dynamic process subdivided in three steps: initiation (step1), linear contraction (step2) and clot stabilization (step3), with a specific involvement of the different cell compartments in each step: platelets/fibrinogen, platelets/fibrinogen/RBCs, and FXIIIa to trigger crosslinking of fibrin, respectively. Recently FXIII activity was shown to be critical for retention of RBCs within the clot and affecting the size of thrombus in healthy subjects [152] and stabilizing the clot in SCD patients [153]. Tutwiler et al [28] also reported an inverse correlation between hematocrit (Hct) values and the extent of clot contraction with healthy blood. However, the same experiment performed with sickle blood showed opposite results. Indeed, despite the lower Hct values usually observed in SCD patients compared to healthy subjects, the extent of contraction is lower in the former. Further studies are needed to understand these differences between healthy and SCD patients.
Complications associated with a hypercoagulable state in SCD patients
Thrombosis-related complications of SCD such as stroke, silent cerebral infarct, and VTE have recently been described in depth by Noubouossie et al [86]. Thus, SCD complications associated with this hypercoagulable state and their prevalence will be only briefly summarized herein.
Cerebrovascular diseases including acute ischemic stroke, hemorrhagic stroke, and silent cerebral infarction were reported as the most frequent complications in children with SCD [154]. Elevated transcranial Doppler ultrasound velocity (TCD) (>200 cm/s) is a marker of high ischemic stroke risk in SCD children [155]. Moreover, several studies found that lower TCD velocity in SCD children correlated with lower serum levels of biomarkers of coagulation (TAT complex, D-dimer, Von Willebrand Factor) [156] and lower thrombin generation in platelet poor plasma [157]. Children with a history of stroke were also reported to have elevated levels of RBC- and platelet-derived MPs [147]. The risk of stroke was also reported to increase with age in SCD patients with 310/100,000 person-years in children (<18 years old) versus 4700/100,000 person-years in adults aged 65 years or older [158]. An association between the history of stroke in SCD adult patients and D-dimer levels was also reported [5]. Few studies reported contrasting results, which could be linked to the way cerebrovascular disease was assayed [108] or the time of steady state versus VOC [159]. Nevertheless, overall results indicate a possible role of hypercoagulation in SCD-associated stroke in children and adults. Strokes can be classified in three specific subgroups more or less common according to the age of the patients [160]. Ischemic stroke occurred most frequently in young children and in patients 30 years and older [154], while hemorrhagic strokes were most frequently reported in young adults [161]. The third type includes silent cerebral infarcts (SCI), which are probably the most frequent cerebral injury in children and adults with SCD [162, 163] occurring in 37.1% of the SCD population below 14 years old [164]. SCI increases the risk of overt stroke and cognitive impairment [162]. Thus the recognition and the early diagnosis of SCD patients at risk of stroke and SCI may be a critical point for the management of SCD patients [160]. In this way, a recent review [165] described promising neuro-biomarkers that could be used to prevent or treat neurological complications in SCD children.
Furthermore, another thrombotic complication, VTE, is now a recognized complication of SCD [22, 23] with a high prevalence in hospitalized SCD patients [25, 166]. Although data are scarce, a recent review also reported a higher risk for VTE in children with SCD [167]. A most recent retrospective cohort study with up to 23 years of follow-up reported that hospitalization frequency did not correlate with elevated risk of VTE in SCD patients [22]. Indeed, almost half of the VTE events occurred 90 days after hospital discharge and the incidence of VTE in African American SCD patients was significantly higher compared to race-matched hospitalized asthma controls. Others factors such as pregnancy [168, 169], catheter use [170, 171], higher baseline levels of hemoglobin, and surgical splenectomy [172] may also affect the risk of VTE in SCD patients. Among VTE, pulmonary embolism (PE) was reported to be a more frequent complication compared to deep venous thrombosis (DVT) [23, 25]. Indeed, around a 4-fold higher incidence of pulmonary embolism (PE) was found in hospitalized patients with SCD compared with non-SCD African American patients at the same age (<40 years old) [25] whereas no differences were found for DVT (DVT). Those results are consistent with previous studies that reported PE in autopsied patients with SCD [173, 174] but differ from another study [175] in which the prevalence of PE in SCD patients was similar to those observed in age and race-matched non-SCD patients. Under-diagnosed PE in SCD patients due to under-utilization of computerized tomographic scans may explain those results [175]. In addition to this higher incidence of VTE in patients with SCD, Brunson et al [22] showed that the first VTE event occurred at a younger age (around 31 years old) in SCD patients than the first VTE event in the general population, and carries a higher risk of mortality [22, 23].
Conclusion
The chronic activation of coagulation is now well recognized as part of the pathophysiology of SCD. The higher levels of TF expression, PS exposure, and NETs are potent activators of the coagulation cascade. Specific properties of sickle RBCs associated with chronic hemolysis and subsequent release of free heme and MPs partly drive this process. Meanwhile, activated coagulation may not only trigger thrombotic complications but also enhance chronic inflammation and the occurrence of VOC. Thus, future clinical studies should investigate the effects of new anticoagulant therapies on the complex pathophysiology of SCD that preferably have minimal impact on hemostasis [176]. In that context, targeting components of the intrinsic pathway could eliminate the risk of serious hemorrhage yet still provide benefits associated with reduced thrombosis, vascular inflammation, and end-organ damage in SCD [176, 177].
Footnotes
Acknowledgments
We would like to thank Drs. Raj Kasthuri and Brandi Reeves for their helpful comments.