
Abstract
The aim of the present study was to compare blood rheological parameters between children with homozygous sickle cell disease (SS), sickle cell SC disease or S/β+-thalassemia syndrome, and healthy children (AA) and to test the associations between blood rheology and the clinical severity in S/β+-thalassemia. Sixty-two SS, 14 SC, 11 S/β+-thalassemia and 12 healthy children participated in this study. Blood viscosity was measured with a cone-plate viscometer at 225 s–1. Red blood cell (RBC) deformability was measured by ektacytometry and RBC aggregation, by syllectometry. Nitric oxide and nitrotyrosine levels were determined for each child. While most of the hematological parameters were not different between SC and S/β+-thalassemia children, we demonstrated that SC patients had lower RBC deformability and aggregation than S/β+ individuals. Nitrotyrosine level, which indicates peroxynitrite production, was similar and lower in both healthy and S/β+ compared to SS children. However, S/β+-thalassemia children who experienced vaso-occlusive crises (VOC) in the 2 previous years had lower NOx and higher nitrotyrosine levels than those who never had VOC within the same period. These findings suggest that vascular function could be impaired in the most severe S/β+-thalassemia children compared to the less severe one.
Introduction
Sickle cell disease (SCD) is a severe monogenic hemoglobinopathy characterized by the synthesis of an abnormal hemoglobin (HbS). In addition to the homozygous form (SS), SCD can also result from compound heterozygosity associating HbS with HbC, or β-thalassemia mutation (decrease (β+) or absence (β0) of β-globin synthesis). The clinical picture of individuals with S/β+-thalassemia syndrome is usually considered less severe than the one of SS patients (including SS or S/β0 patients) or SC patients (patients with both HbS and HbC) [6]. However, while some S/β+-patients can be asymptomatic, others may develop severe complications, as it is the case for SS and SC patients [2]. The type of mutation at the origin of β+-thalassemia syndrome plays an important role in the modulation of the clinical severity by modulating the amount of HbA into the red blood cells (RBC) [2, 9].
Several studies demonstrated that blood rheology is severely impaired in SS and SC patients [10, 34]. Because anemia is more important in SS than in SC patients, blood viscosity is usually lower in the former group [34]. However, any increase in blood viscosity in SS patients may trigger painful vaso-occlusive crisis [8, 19]. Both SS and SC patients have decreased RBC deformability compared to normal individuals but the magnitude of the alterations is higher in SS individuals [34, 37]. Few studies also investigated RBC aggregation in SS and SC patients and reported increased RBC aggregates strength compared to controls [19, 37]. While the blood rheological profiles of SS and SC patients are now pretty well described, no study investigated the role of blood rheology in the pathophysiology of S/β+-thalassemia syndrome. The aim of the present study was to compare blood rheological parameters between children with SS, SC or S/β+-thalassemia syndrome, and healthy children (AA) and to test the associations between blood rheology and the clinical severity in S/β+-thalassemia.
Material and methods
Patients
Sixty-two SS, 14 SC and 11 S/β+-thalassemia children were recruited at the Oncologic and Hematologic Pediatric Institute of Lyon (Hospices Civils de Lyon, France) from January 2014 to January 2016. Twelve healthy ethnic-matched children were also recruited as control group. SCD diagnosis was performed using three complementary biochemical tests according to expert recommendations [1] and genetically confirmed thereafter. All patients were at steady state at the time of the study (i.e., no blood transfusion within the last three months, absence of acute episodes of infection, vaso-occlusive crisis (VOC) or acute chest syndrome (ACS) at least one month before inclusion in the study). The number of hospitalized VOC and ACS was recorded over a 2 yrs period before the date of inclusion. VOC and ACS were defined as previously published [19, 32]. All children and their parents were informed about the purpose and procedures of the study, which was approved by the “Hospices Civils de Lyon — CPP Est” (ethics committee number: L14-127) and conducted in accordance to the Declaration of Helsinki.
Biological parameters
Venipuncture was performed between 7:00 and 10:00 a.m., and blood samples were used immediately for analyses. Hemoglobin concentration (Hb), hematocrit (Hct), percent reticulocytes (RET), mean cell volume (MCV) and Mean Corpuscular Hemoglobin Concentration (MCHC) were determined using a hematology analyzer (Advia, Siemens, Rungis, France).
Measurements of hemolytic markers (bilirubin, BIL; lactate dehydrogenase, LDH; aspartate aminotransferase, AST) were performed using standard biochemistry in SS, SC and S/β+-thalassemia children. NO end products (NOx) were measured using the Griess method [27]. Nitrate reductase and Nicotinamide adenine dinucleotide phosphatase (NADPH) were added to plasma in order to reduce nitrate to nitrite. Concentrations of plasma nitrotyrosine, as end product of protein nitration induced by peroxynitrite, were measured using a competitive ELISA test as previously described [13].
Blood viscosity was determined at native Hct and a shear rate of 225 s–1 at 25°C, using a cone/plate viscometer (Brookfield DVII+ with CPE40 spindle, Brookfield Engineering Labs, Natick, MA), and expressed in centipoises (cP). RBC deformability was determined at 37°C at 3 and 30 Pa by laser diffraction analysis (ektacytometry), using the Laser-assisted Optical Rotational Cell Analyzer (LORRCA Maxsis, RR Mechatronics, Hoorn, The Netherlands). RBC aggregation was determined at 37°C via syllectometry, (i.e., laser backscatter versus time, using the LORRCA) after adjustment of the Hct to 40%, and reported as the aggregation index (AI). RBC aggregates strength, i.e., the minimal shear rate needed to prevent RBC aggregation or to breakdown existing RBC aggregates, was determined using a re-iteration procedure [15] using the LORRCA. The guidelines for international standardization in blood rheology techniques/measurements [4], as well as for hemorheological measurements in sickle cell disease [33], were strictly followed.
Statistics
Data are expressed as means±standard deviation. A one-way ANOVA was used with LSD post-hoc test to compare the different parameters between the four groups. Then, the S/β+-thalassemia group was divided according to either 1) the positive or negative history for hospitalized painful vaso-occlusive crises within the 2 previous years or 2) the percentage of hemoglobin A (<15% vs >15%). An unpaired Student t-test was used to compare the S/β+-thalassemia subgroups. The significance level was defined as p < 0.05 (SPSS, v. 20, IBM SPSS Statistics, Chicago, IL).
Results
Molecular studies
Among the 11 S/β+-thalassemia children, 4 different mutations were detected associated with HbS mutation: the HbE mutation (HBB:c.79G>A) in 5 patients, the HBB:c.92+5G>T mutation in 1 patient, the HBB:c.-79A>G mutation in 2 patients and the HBB:c.-138C>T in 3 patients. The HbE variant is one of the most frequent variants after HbS and HbC. It is especially represented in Southeast Asia with a prevalence of 30–45% [35]. The HbE mutation creates an alternative splice donor site which decreases functional bêtaE globin synthesis [31]. The HBB:c.92+5G>T mutation, which is also particularly frequent in Southeast Asia [17], reduces the splicing efficiency of the splice donor site and is thus considered as a severe β+-thalassemia mutation with a low HbA level besides HbS [14, 36]. The HBB:c.-79A>G and HBB:c.-138C>T mutations decrease HBB gene transcription because of a reduced binding of transcription factors [16, 30].
Clinical characteristics and hematology
The values are reported in the Table 1. As expected, SS children had higher number of ACS over the 2 yrs period than SC and S/β+-thalassemic children. Although there was no statistical difference in the VOC mean value between the groups, SS children had clinically more episodes than the two other sickle cell groups. About one third of SS children was treated by hydroxurea to decrease the frequency of vaso-occlusive events [7, 24] while no SC or S/β+-thalassemia children took this medication. Compared to AA children, Hct and Hb were lower in the three SCD groups but anemia was more pronounced in SS children. Hemolytic markers (RET, BIL, LDH and RET) were higher in SS than in SC and S/β+-thalassemia children. The MCV of the 3 SCD groups was lower than in AA children but both SC and S/β+-thalassemia individuals had lower MCV than SS patients. In addition, S/β+-thalassemia children had lower MCV than the SC group. MCHC of the SC group was higher than S/β+-thalassemia and SS children.
General characteristics and hematological parameters in SS, SC, S/β+-thalassemia and AA children
General characteristics and hematological parameters in SS, SC, S/β+-thalassemia and AA children
HU, hydroxyurea treated patients; VOC, vaso-occlusive crises; ACS, acute chest syndrome; Hb, hemoglobin concentration; Hct, hematocrit; MCV, mean cell volume; MCHC, mean corpuscular hemoglobin concentration; RET, reticulocytes; BIL, bilurubine; AST, aspartate aminostransferase; LDH, lactate dehydrogenase. Different from SS: *p < 0.05; **p < 0.01; ***p < 0.001; different from SC: $p < 0.05; $$p < 0.01; $$$p < 0.001; different from S/β+-thalassemia: ‡p < 0.05; ‡‡p < 0.01; ‡‡‡p < 0.001.
The values of the four groups are reported in Table 2. Blood viscosity was lower in SS than in SC, S/β+-thalassemia and AA children. RBC deformability at 3 Pa was not different between SS and SC children while, at 30 Pa, the deformability of RBC was lower in SS than in SC children. Children with S/β+-thalassemia had more deformable RBCs than SS and SC groups at 3 Pa. At 30 Pa, S/β+-thalassemia group still have higher RBC deformability than SS children but only a trend was noted with the SC group (p < 0.1). RBC deformability was lower in the three sickle cell groups in comparison with AA children. RBC aggregation was lower in SC children compared to the three other groups and was lower in SS children compared to AA individuals. However, no difference was observed between the AA and S/β+-thalassemia groups. RBC aggregates strength was higher in both SS and SC groups compared to AA children, and higher in SS than in S/β+-thalassemic children. No difference in the RBC aggregates strength was observed between children with S/β+-thalassemia and healthy children. NOx was higher in the three SCD groups compared to AA children and lower in SC than in SS patients. Nitrotyrosine level was increased in SS children compared to S/β+-thalassemia and healthy children. The nitrotyrosine mean value of the SC group was comprised between SS and S/β+-thalassemia values.
Hemorheological parameters, nitric oxide and nitrotyrosine levels in SS, SC, S/β+-thalassemic and AA children
Hemorheological parameters, nitric oxide and nitrotyrosine levels in SS, SC, S/β+-thalassemic and AA children
RBC, red blood cell. Different from SS: *p < 0.05; **p < 0.01; ***p < 0.001; different from SC: $p < 0.05; $$p < 0.01; $$$p < 0.001; different from S/β+-thalassemia: ‡p < 0.05; ‡‡p < 0.01; ‡‡‡p < 0.001.
Five S/β+-thalassemia children were hospitalized for painful episodes during the two years period preceding the study (VOC group; number of events ranging from 1 to 5) while the 6 other presented no VOC during that time. Comparison between these two subgroups (Fig. 1) demonstrated lower Hb, NOx and blood viscosity in the VOC group. In contrast, nitrotyrosine level tended to be higher in the VOC compared to the no-VOC group. The other blood rheological and hematological parameters were not significantly different between the two groups (data not shown). We also divided the S/β+-thalassemia children according to their HbA percentage (<15% versus ≥15%) but no significant difference was observed between the two subgroups (data not shown). No association was observed between RBC deformability and the HbA percentage (r = 0.39, p = 0.34 at 3 Pa; r = 0.04, p = 0.92 at 30 Pa), Hb (r = 0.17; p = 0.61) or LDH (r = 0.33; p = 0.33).
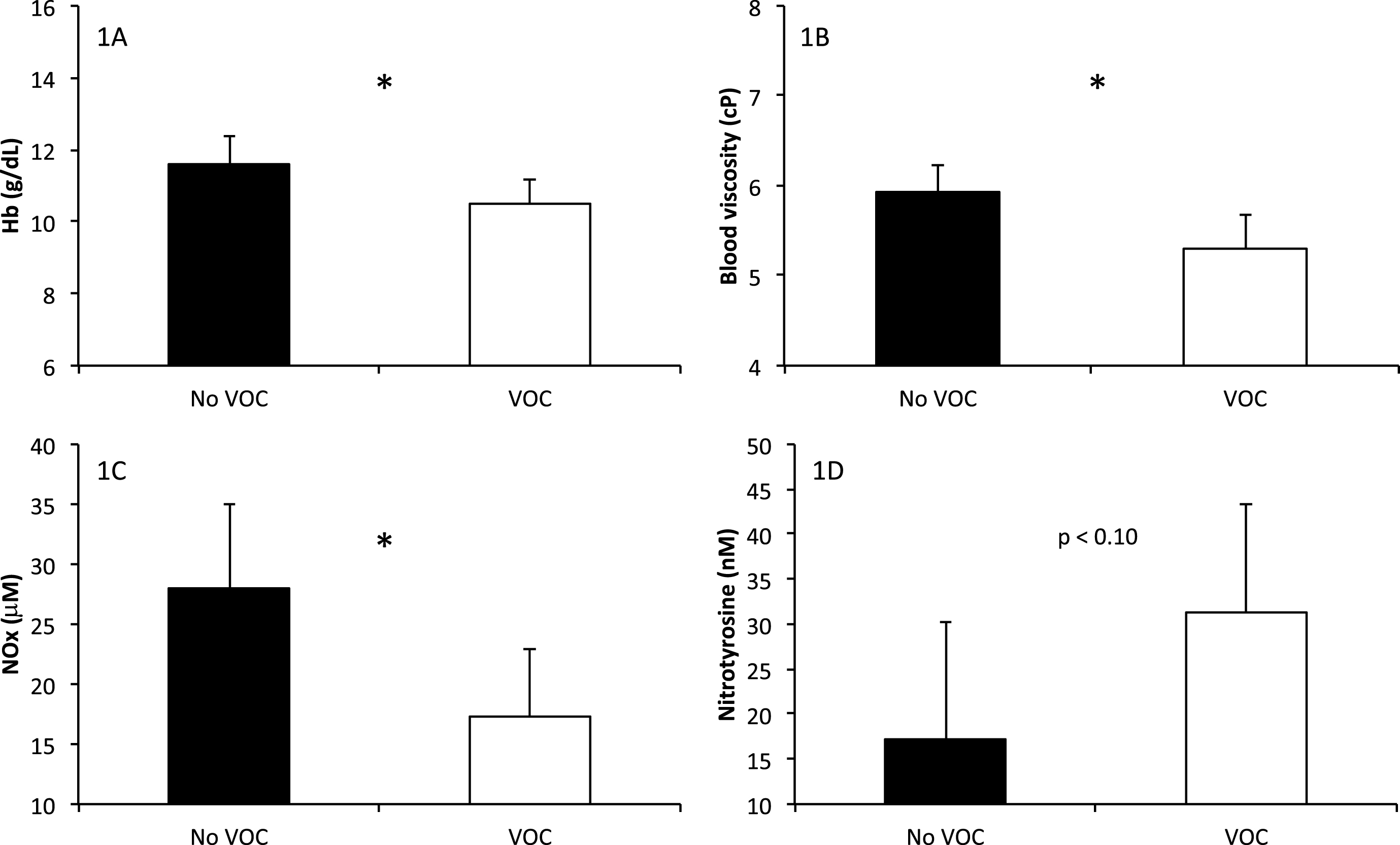
Biological parameters in S/β+-thalassemia categorized according to the positive history of hospitalized vaso-occlusive crises or not within the 2 yrs preceding the study: 1A) hemoglobin concentration, 1B) blood viscosity, 1C) nitric oxide end-products, 1D) nitrotyrosine, which reflects peroxynitrite production. Significant difference: *p < 0.05.
The present study compared several hematological and hemorheological parameters between SS, SC, S/β+-thalassemia and healthy children. While most of the hematological parameters are not different between SC and S/β+-thalassemia children, the blood rheology study demonstrated that SC patients have lower RBC deformability and aggregation than S/β+ individuals. Nitrotyrosine level, which indicates peroxynitrite production, is similar and lower in both healthy and S/β+ compared to SS children. However, S/β+-thalassemia children who experienced VOC had lower NOx and higher nitrotyrosine levels compared to those who did not have VOC.
Studies in the field of SCD usually separate SS and SC children because these two genotypes are characterized by different frequencies of complications [19, 26]. SS and SC children may both experience frequent VOC but the former population is at greater risk for stroke, pulmonary hypertension, priapism or leg ulcers [5, 26]. In contrast, SC individuals are more prone to develop retinopathy, otologic disorders and osteonecrosis, mainly at adult age [26]. S/β+-thalassemia is considered as a mild sickle cell disorder with less frequent clinical complication [6]. Nevertheless, a significant proportion of S/β+ patients may exhibit frequent vaso-occlusive complications and the reasons are not completely understood [6].
Our study confirmed that SS disease is more severe than the other sickle cell syndromes with more frequent vaso-occlusive like complications. As it is the case in the present study, severe anemia is responsible for a decrease of blood viscosity in SS population compared to healthy individuals and the other sickle cell syndromes. However, any increase in blood viscosity may trigger vaso-occlusive crisis in SS patients [8, 19]. The main reason is that chronic hemolysis interferes with the metabolism of nitric oxide leading to a loss of vascular reactivity [18]. Indeed, the vascular system of SS individuals is not able to cope with the increase of blood viscosity, resulting in clinical complications [8]. SC and S/β+-thalassemia children have a mild anemia compared to SS children, which may explain why blood viscosity is higher in these two populations compared to SS individuals.
Blood viscosity depends on RBC biophysical properties [4]. While RBC deformability was not different between SS and SC children at 3 Pa, it was reduced in SS children at 30 Pa compared to SC, S/β+-thalassemia and healthy children. This severe RBC deformability reduction in SS population is involved in both the pathophysiology of several clinical complications [10] and the enhanced fragility of RBCs [11].
RBC deformability was also reduced in SC children in comparison with healthy and S/β+-thalassemia children. HbC has been demonstrated to promote RBC dehydration in SC individuals, thus increasing HbS polymerization and RBC sickling [3, 12]. The increased MCHC observed in SC compared to S/β+-thalassemia children supports the fact that RBCs from SC children are more dehydrated than in S/β+-thalassemia. This decreased RBC deformability in SC population, associated with an increased RBC aggregates stickiness, could explain the increased risk to develop ischemic-like complications [22]. Although RBC deformability was better preserved in S/β+-thalassemia children, it was also reduced compared to the AA group but the degree of the reduction did not explain the clinical variability of the children (no difference between the VOC and no VOC groups) and was not related to the percentage of hemoglobin A.
Although of limited sample size, the S/β+-thalassemia group was composed of two subgroups of children: those who exhibited VOC in the 2 previous years before the study and those who did not. The slightly higher anemia was at the origin of the lower blood viscosity found in the VOC group. This finding is intriguing since decreased blood viscosity theoretically protects from frequent vaso-occlusive like episodes, as it is the case in SS population [8, 29]. However, we also observed that S/β+-thalassemia children with VOC had lower NOx level and higher peroxynitrite production than their VOC free counterpart, suggesting the presence of higher amount of reactive oxygen species, which decreased nitric oxide bioavailibity in the most severe subgroup. On the whole, S/β+-thalassemia children had, like SS children, higher NOx level than healthy children. The lower peroxynitrite production in S/β+-thalassemia children compared to SS, suggest a more preserved nitric oxide bioavailability. However, our findings suggest that the most severe S/β+-thalassemia children have impaired nitric oxide bioavailability, which could participate to increase the risks for VOC events. Further studies are needed to carefully investigate the vascular function of children with S/β+-thalassemia to address its role in the clinical modulation of this phenotype.
In conclusion, the hematological profile of SC and S/β+-thalassemia children is rather similar. However, the reduction of RBC deformability is less severe in S/β+-thalassemia individuals than in SC patients. The slight reduction in RBC deformability observed in S/β+-thalassemia children was not associated with the clinical severity. In contrast, S/β+-thalassemia children with the lowest NOx level and higher peroxynitrite production could be at greater risk to develop vaso-occlusive like complications.